Problématiques Environnementales Et Du Développement Durable
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

No 1223/2009 of the EUROPEAN PARLIAMENT and of the COUNCIL of 30 November 2009 on Cosmetic Products (Recast) (Text with EEA Relevance) (OJ L 342, 22.12.2009, P
02009R1223 — EN — 03.12.2020 — 025.001 — 1 This text is meant purely as a documentation tool and has no legal effect. The Union's institutions do not assume any liability for its contents. The authentic versions of the relevant acts, including their preambles, are those published in the Official Journal of the European Union and available in EUR-Lex. Those official texts are directly accessible through the links embedded in this document ►B REGULATION (EC) No 1223/2009 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 30 November 2009 on cosmetic products (recast) (Text with EEA relevance) (OJ L 342, 22.12.2009, p. 59) Amended by: Official Journal No page date ►M1 Commission Regulation (EU) No 344/2013 of 4 April 2013 L 114 1 25.4.2013 ►M2 Commission Regulation (EU) No 483/2013 of 24 May 2013 L 139 8 25.5.2013 ►M3 Commission Regulation (EU) No 658/2013 of 10 July 2013 L 190 38 11.7.2013 ►M4 Commission Regulation (EU) No 1197/2013 of 25 November 2013 L 315 34 26.11.2013 ►M5 Commission Regulation (EU) No 358/2014 of 9 April 2014 L 107 5 10.4.2014 ►M6 Commission Regulation (EU) No 866/2014 of 8 August 2014 L 238 3 9.8.2014 ►M7 Commission Regulation (EU) No 1003/2014 of 18 September 2014 L 282 1 26.9.2014 ►M8 Commission Regulation (EU) No 1004/2014 of 18 September 2014 L 282 5 26.9.2014 ►M9 Commission Regulation (EU) 2015/1190 of 20 July 2015 L 193 115 21.7.2015 ►M10 Commission Regulation (EU) 2015/1298 of 28 July 2015 L 199 22 29.7.2015 ►M11 Commission Regulation (EU) 2016/314 of 4 March 2016 L 60 59 5.3.2016 ►M12 Commission Regulation (EU) -

List of Lists
United States Office of Solid Waste EPA 550-B-10-001 Environmental Protection and Emergency Response May 2010 Agency www.epa.gov/emergencies LIST OF LISTS Consolidated List of Chemicals Subject to the Emergency Planning and Community Right- To-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act • EPCRA Section 302 Extremely Hazardous Substances • CERCLA Hazardous Substances • EPCRA Section 313 Toxic Chemicals • CAA 112(r) Regulated Chemicals For Accidental Release Prevention Office of Emergency Management This page intentionally left blank. TABLE OF CONTENTS Page Introduction................................................................................................................................................ i List of Lists – Conslidated List of Chemicals (by CAS #) Subject to the Emergency Planning and Community Right-to-Know Act (EPCRA), Comprehensive Environmental Response, Compensation and Liability Act (CERCLA) and Section 112(r) of the Clean Air Act ................................................. 1 Appendix A: Alphabetical Listing of Consolidated List ..................................................................... A-1 Appendix B: Radionuclides Listed Under CERCLA .......................................................................... B-1 Appendix C: RCRA Waste Streams and Unlisted Hazardous Wastes................................................ C-1 This page intentionally left blank. LIST OF LISTS Consolidated List of Chemicals -

Federal Register/Vol. 84, No. 230/Friday, November 29, 2019
Federal Register / Vol. 84, No. 230 / Friday, November 29, 2019 / Proposed Rules 65739 are operated by a government LIBRARY OF CONGRESS 49966 (Sept. 24, 2019). The Office overseeing a population below 50,000. solicited public comments on a broad Of the impacts we estimate accruing U.S. Copyright Office range of subjects concerning the to grantees or eligible entities, all are administration of the new blanket voluntary and related mostly to an 37 CFR Part 210 compulsory license for digital uses of increase in the number of applications [Docket No. 2019–5] musical works that was created by the prepared and submitted annually for MMA, including regulations regarding competitive grant competitions. Music Modernization Act Implementing notices of license, notices of nonblanket Therefore, we do not believe that the Regulations for the Blanket License for activity, usage reports and adjustments, proposed priorities would significantly Digital Uses and Mechanical Licensing information to be included in the impact small entities beyond the Collective: Extension of Comment mechanical licensing collective’s potential for increasing the likelihood of Period database, database usability, their applying for, and receiving, interoperability, and usage restrictions, competitive grants from the Department. AGENCY: U.S. Copyright Office, Library and the handling of confidential of Congress. information. Paperwork Reduction Act ACTION: Notification of inquiry; To ensure that members of the public The proposed priorities do not extension of comment period. have sufficient time to respond, and to contain any information collection ensure that the Office has the benefit of SUMMARY: The U.S. Copyright Office is requirements. a complete record, the Office is extending the deadline for the extending the deadline for the Intergovernmental Review: This submission of written reply comments program is subject to Executive Order submission of written reply comments in response to its September 24, 2019 to no later than 5:00 p.m. -

Cancellations for USP38-NF33
Compendial Cancellations for USP38-NF33 Category Monograph Title Monograph Section Scientific Liaison <111> DESIGN AND ANALYSIS OF BIOLOGICAL ASSAYS PF Revision 39(4) Pg. ONLINE General Maura Kibbey Title, INTRODUCTION, POLYETHYLENE CONTAINERS, POLYPROPYLENE CONTAINERS, POLYETHYLENE TEREPHTHALATE BOTTLES AND POLYETHYLENE TEREPHTHALATE G Revision <661> CONTAINERS -- PLASTICS PF 39(5) Pg. ONLINE CONTAINERS, TEST METHODS, SCOPE Desmond Hunt <661.1> PLASTIC MATERIALS OF CONSTRUCTION PF 39(5) New Pg. ONLINE Title, INTRODUCTION, SCOPE, TEST METHODS, SPECIFICATIONS Desmond Hunt <661.2> PLASTIC PACKAGING SYSTEMS FOR New PHARMACEUTICAL USE PF 39(5) Pg. ONLINE Title, INTRODUCTION, SCOPE, TEST METHODS, SPECIFICATIONS Desmond Hunt ULTRAVIOLET, VISIBLE, INFRARED, ATOMIC ABSORPTION, FLUORESCENCE, TURBIDIMETRY, <851> SPECTROPHOTOMETRY AND LIGHT-SCATTERING PF NEPHELOMETRY, AND RAMAN MEASUREMENTLIGHT-SCATTERING, COMPARATIVE UTILITY OF Revision 39(3) Pg. ONLINE SPECTRAL RANGES, THEORY AND TERMS, APPARATUS, PROCEDURE Horacio Pappa ASSAY/Procedure, IMPURITIES/Organic Impurities, IDENTIFICATION/Infrared Absorption Revision AMANTADINE HYDROCHLORIDE PF 39(3) Pg. ONLINE <197S>, ASSAY/Procedure Shankari Shivaprasad AMANTADINE HYDROCHLORIDE CAPSULES PF 39(5) Pg. Revision ONLINE PERFORMANCE TESTS/Dissolution <711> Shankari Shivaprasad Title, DEFINITION/Introduction, IDENTIFICATION/A., ASSAY/Procedure, PERFORMANCE TESTS/Dissolution <711>, PERFORMANCE TESTS/Uniformity of Dosage Units <905>, IMPURITIES/Procedure for Artemether, IMPURITIES/Procedure for Lumefantrine, -

Ep 0872229 A1
Patentamt Europaisches ||| || 1 1| || || || || || || || || ||| || (19) J European Patent Office Office europeen des brevets (11) EP 0 872 229 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51 ) |nt. CI.6: A61 K 7/06, A61 K 7/48, 21.10.1998 Bulletin 1998/43 ^61 K 7/50 (21) Application number: 97201101.9 (22) Date of filing: 14.04.1997 (84) Designated Contracting States: (72) Inventors: AT BE CH DE DK ES Fl FR GB GR IE IT LI LU MC • Embrechts, Roger Carolus Augusta NL PT SE 2340 Beerse (BE) Designated Extension States: • Odds, Frank Christopher AL LT LV RO SI 2340 Beerse (BE) • de Doncker, Piet R.G. (71) Applicant: 2340 Beerse (BE) JANSSEN PHARMACEUTICA N.V. 2340 Beerse (BE) (54) Compositions containing an antifungal and a phospholipid (57) The invention relates to compositions such as body and hair cleansing products, in particular sham- poos, comprising one or more antifungals inhibiting fun- gal ergosterol biosynthesis as a first active ingredient, a synthetic, amphotheric phospholipid acting both as a second active ingredient and as a surface active agent, and art-known body or hair cleansing product ingredi- ents as a carrier. < CM CM CM co o Q_ LU Printed by Xerox (UK) Business Services 2.16.3/3.4 EP 0 872 229 A1 Description The invention relates to compositions such as body and hair cleansing products, in particular shampoos, compris- ing one or more antifungals inhibiting fungal ergosterol biosynthesis as a first active ingredient, a synthetic, amphotheric 5 phospholipid acting both as a second active ingredient and as a surface active agent, and art-known body or hair cleansing product ingredients as a carrier. -

United States Patent (19) 11 Patent Number: 6,077,832 Chamberlain Et Al
USOO6077832A United States Patent (19) 11 Patent Number: 6,077,832 Chamberlain et al. (45) Date of Patent: *Jun. 20, 2000 54). ANTIVIRAL BENZIMIDAZOLE OTHER PUBLICATIONS NUCLEOSIDE ANALOGUES AND A METHOD FOR THEIR PREPARATION Methods of Nucleoside Synthesis. Vorbrueggen, Helmut. Res. Lab., Schering A-G., Berlin, D-1000/65, Fed. Rep. 75 Inventors: Stanley Dawes Chamberlain; Susan Ger. NATO Adv. Study Inst. Ser, Ser. A (1979), Mary Daluge; George Walter A26(Nucleoside Analogues: Chem., Biol., Med. Appl.), Koszalka, all of Chapel Hill, N.C. 35-69. Vorbriggen et al., “Nucleoside Synthesis with Trimethylsi 73 Assignee: GlaxoWellcome Inc., Five Moore lyl Triflate and Perchlorate as Catalysts,” Chem. Ber. 114, Drive, N.C. pp. 1234–1255 (1981). Vorbriggen et al., “New Catalysts for the Synthesis of * Notice: This patent issued on a continued pros Nucleosides,” Angew. Chem. Internat. Edit. 14(6), pp. ecution application filed under 37 CFR 421–422 (1974). 1.53(d), and is subject to the twenty year Gordon et al., “Kinetics of Decay in the Expression of patent term provisions of 35 U.S.C. Interferon-Dependent mRNAS Responsible for Resistance 154(a)(2). to Virus.” Proc. Nat. Acad. Sci. USA 77(1), 452–456 (Jan. 21 Appl. No.: 08/765,758 1980). Devivar et al., “Benzimidazole Ribonucleosides: Observa 22 PCT Filed: Jul. 6, 1995 tion of An Unexpected Nitration WHen Performing Non Aqueous Diazotizations with t-butyl Nitrite,” Biorganic C& 86 PCT No.: PCT/GB95/01597 Medicinal Chem. Letters, 2(9), 1105–1110 (Sep. 1992). S371 Date: Jan. 6, 1997 Tigges et al., “Human CD8+ Herpes Simplex Virus-Specific Cytotoxic T-Lymphocyte Clones Recognize Diverse Viron S 102(e) Date: Jan. -

Flash Environmental Assessment Tool (Feat) Version 2.0
FLASH ENVIRONMENTAL ASSESSMENT TOOL (FEAT) VERSION 2.0 Reference Guide Working Document To identify acute environmental risks immediately following disasters DISCLAIMER FEAT combines large amounts of scientific insights and data into one simple tool for use in field-based situations. Assumptions are made in the FEAT, some of them approximate. FEAT was prepared as an account of work sponsored by the United Nations and the Dutch National Institute for Public Health and the Environment. Readers and users of FEAT are responsible for their operations and functions. FEAT outputs will help to prioritize activities of relief and risk management teams, but cannot provide definitive scientific assessments or analysis. For example, FEAT cannot provide exact impact perimeters. Exact results will depend on individual cases and conditions. Users will need to set priorities based on actual field situations, which may differ from those presented in this document. FEAT is intended as guidance material only, and is not to be regarded as binding international regulation or legislation. FEAT does not reflect nor replace nor supersede national, regional or international legislation, regulations or policies. FEAT data has been derived from recognized scientific databanks of international authorities or institutions in good faith and on basis of information available at the date of publication. Resources include amongst others the European Chemicals Agency, UNECE Industrial Accidents Convention Directives and the US Environmental Protection Agency. Neither the UN nor any of its agencies or employees make any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or any third party's use or the results of such use of any information. -

Antifungal Drugs Are Those Drugs Which Inhibit Or Retard Fungal Growth
Prepared by- Dr Arpita Shrivastav DR ARPITA SHRIVASTAV ASSTT. PROFESSOR VETERINARY COLLEGE REWA Antifungal drugs are those drugs which inhibit or retard fungal growth. DR ARPITA SHRIVASTAV ASSTT. PROFESSOR VETERINARY COLLEGE REWA Sites of action of antifungal drugs Chitin + Glucan DR ARPITA SHRIVASTAV ASSTT. PROFESSOR VETERINARY COLLEGE REWA SITE OF ACTION DRUGS Cell membrane Ergosterol binding Amphotericin B, Natamycin and Nystatin Ergosterole synthesis inhibitors Allylamines (Amorolfine, (Squalene epoxidase inhibitor) Terbinafine etc.) Ergosterol synthesis inhibitors Azoles (Ketoconazole, Miconazole (lanosterol to ergosterol) etc.) Cell wall β glucan synthase inhibitor Echinocandins Intracelluler Pyrimidine analogue Flucytosine Mitotic inhibitors Gresiofulvin Other site Benzoic acid , salicylic acid, tolnaftate, castor oil etc. DR ARPITA SHRIVASTAV ASSTT. PROFESSOR VETERINARY COLLEGE REWA Source – Streptomyces nodosus Mechanism of action – Increases permeability of cell membrane by binding to ergosterol Antimicrobial spectrum – broad spectrum Resistance – when less ergosterol in membrane Pharmacokinetics – poorly absorbed from GIT, do not cross BBB Adverse effects - Nephrotoxicity Drug interaction – 1. with miconazole antagonistic effect 2. with flucytosine synergistic effect Dose – Dog -0.25 to 0.50 mg/kg, 3 times weekly Cat – 0.1 to 0.50 mg/kg, 3 times weekly DR ARPITA SHRIVASTAV ASSTT. PROFESSOR VETERINARY COLLEGE REWA DR ARPITA SHRIVASTAV ASSTT. PROFESSOR VETERINARY COLLEGE REWA Source – Streptomyces noursie Streptomyces aureus Mechanism of action – It binds to ergosteol and forms pores in cell membrane DR ARPITA SHRIVASTAV ASSTT. PROFESSOR VETERINARY COLLEGE REWA Source - Streptomyces natalensis Mechanism of action – Binds to ergosterol and cause leakiness of cell membrane DR ARPITA SHRIVASTAV ASSTT. PROFESSOR VETERINARY COLLEGE REWA Pentameric ring compounds containing at least one other non-carbon atom of either nitrogen, sulfur, or oxygen. -

Table 4: Protective Action Criteria (PAC) Rev. 29 Based on Applicable
Table 4: Protective Action Criteria (PAC) Rev. 29a based on applicable 60-minute AEGLs, ERPGs, or TEELs. The chemicals are 3 listed in alphabetical order and the values are presented in mg/m . June 2018 Table 4 is an alphabetical list of the chemical substances and their corresponding PAC values in mass per unit volume (mg/m3). The conversion of ppm to mg/m3 was carried out assuming normal temperature and pressure, 25°C and 760 mm Hg. The columns presented in Table 4 provide the following information: Heading Definition No. The ordered numbering of the chemicals as they appear in this alphabetical listing Chemical Name The name of the chemical substance submitted to the PAC development team CASRN The Chemical Abstracts Service Registry Number1 for this chemical PAC-1 Based on the applicable AEGL-1, ERPG-1, or TEEL-1 value PAC-2 Based on the applicable AEGL-2, ERPG-2, or TEEL-2 value PAC-3 Based on the applicable AEGL-3, ERPG-3, or TEEL-3 value Chemicals for which AEGLs are available have their chemical name, CASRN, and AEGL values displayed in a bolded and larger font. Chemicals for which ERPGs are available, but not AEGLs, have their chemical name, CASRN, and ERPG values displayed in a bolded font. Chemicals for which TEELs are available, but no AEGLs or ERPGs, have their chemical name, CASRN, and values displayed using a regular font. Additional information on PAC values and TEEL values and links to other sources of information is provided on the Subcommittee on Consequence Assessment and Protective Actions (SCAPA) webpage at http://orise.orau.gov/emi/scapa/default.htm. -
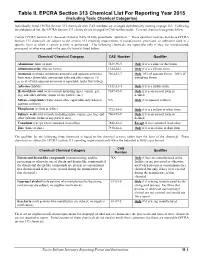
Individually Listed EPCRA Section 313 Chemicals with CAS Numbers Are Arranged Alphabetically Starting on Page II-3
Table II. EPCRA Section 313 Chemical List For Reporting Year 2015 (including Toxic Chemical Categories) Individually listed EPCRA Section 313 chemicals with CAS numbers are arranged alphabetically starting on page II-3. Following the alphabetical list, the EPCRA Section 313 chemicals are arranged in CAS number order. Covered chemical categories follow. Certain EPCRA Section 313 chemicals listed in Table II have parenthetic “qualifiers.” These qualifiers indicate that these EPCRA Section 313 chemicals are subject to the section 313 reporting requirements if manufactured, processed, or otherwise used in a specific form or when a certain activity is performed. The following chemicals are reportable only if they are manufactured, processed, or otherwise used in the specific form(s) listed below: Chemical/ Chemical Category CAS Number Qualifier Aluminum (fume or dust) 7429-90-5 Only if it is a fume or dust form. Aluminum oxide (fibrous forms) 1344-28-1 Only if it is a fibrous form. Ammonia (includes anhydrous ammonia and aqueous ammonia 7664-41-7 Only 10% of aqueous forms. 100% of from water dissociable ammonium salts and other sources; 10 anhydrous forms. percent of total aqueous ammonia is reportable under this listing) Asbestos (friable) 1332-21-4 Only if it is a friable form. Hydrochloric acid (acid aerosols including mists, vapors, gas, 7647-01-0 Only if it is an aerosol form as fog, and other airborne forms of any particle size) defined. Nitrate compounds (water dissociable; reportable only when in NA Only if in aqueous solution aqueous solution) Phosphorus (yellow or white) 7723-14-0 Only if it is a yellow or white form. -

Allyl Acetate CAS # 591-87-7
SUMMARY OF DATA FOR CHEMICAL SELECTION Allyl Acetate CAS # 591-87-7 and Allyl Alcohol CAS # 107-18-6 (Prepared for NCI by Technical Resources, Inc. under contract - (8/91: revised 2/93)) NTP NOMINATION HISTORY AND REVIEW Nomination History • Nomination Source: National Cancer Institute Recommendations: • Metabolism • Carcinogenicity Rationale/Remarks: • Allyl acetate and allyl alcohol were nominated as a pair of chemicals • High production • Potential for high occupational and consumer exposure based on its extensive use in organic synthesis, agriculture and flavoring agents • Mutagenic in Salmonella and mouse lymphoma assays • Available carcinogenicity studies in rats and hamsters were negative but these studies were inadequate (insufficient number of doses, and animals in rat and hamster studies; only one sex used in hamster study) • Higher priority for carcinogenicity testing should be assigned to allyl acetate; if allyl acetate proves to be carcinogenic, priority for carcinogenicity testing of allyl alcohol should be increased Priority: High • Date of Nomination: 4/1993 Table of Contents 1. Chemical Identification a. Basis of Nomination to the CSWG b. Input from Government Agencies/Industry c. Selection Status 2. Exposure Information 3. Evidence for Possible Carcinogenic Activity 4. References CHEMICAL IDENTIFICATION Allyl Acetate CAS Registry Number: 591-87-7 Chem. Abstracts Name: Acetic acid, 2-propenyl ester (9CI); Acetic acid, allyl ester (8CI) Synonyms and Trade Names: Allyl alcohol, acetate; 3-acetoxy-1-propene; 3-acetoxy- propene Allyl Alcohol CAS Registry Name: 107-18-6 Chemical Abstracts Name: 2-Propen-1-o1 (9CI) Synonyms and Trade Names: Allylic alcohol; 3-hydroxypropene; 1-propenol-3; 2-propenol; 2-propenyl alcohol; Shell unkrautted A; Weed Drench; vinyl carbinol Structure, Molecular Formula and Molecular Weight: Allyl Acetate C5H8O2 Mol.