Application of Silicon-Based Cross- Coupling Technology to Aryl Triflates
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
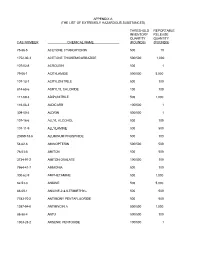
The List of Extremely Hazardous Substances)
APPENDIX A (THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES) THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 75-86-5 ACETONE CYANOHYDRIN 500 10 1752-30-3 ACETONE THIOSEMICARBAZIDE 500/500 1,000 107-02-8 ACROLEIN 500 1 79-06-1 ACRYLAMIDE 500/500 5,000 107-13-1 ACRYLONITRILE 500 100 814-68-6 ACRYLYL CHLORIDE 100 100 111-69-3 ADIPONITRILE 500 1,000 116-06-3 ALDICARB 100/500 1 309-00-2 ALDRIN 500/500 1 107-18-6 ALLYL ALCOHOL 500 100 107-11-9 ALLYLAMINE 500 500 20859-73-8 ALUMINUM PHOSPHIDE 500 100 54-62-6 AMINOPTERIN 500/500 500 78-53-5 AMITON 500 500 3734-97-2 AMITON OXALATE 100/500 100 7664-41-7 AMMONIA 500 100 300-62-9 AMPHETAMINE 500 1,000 62-53-3 ANILINE 500 5,000 88-05-1 ANILINE,2,4,6-TRIMETHYL- 500 500 7783-70-2 ANTIMONY PENTAFLUORIDE 500 500 1397-94-0 ANTIMYCIN A 500/500 1,000 86-88-4 ANTU 500/500 100 1303-28-2 ARSENIC PENTOXIDE 100/500 1 THRESHOLD REPORTABLE INVENTORY RELEASE QUANTITY QUANTITY CAS NUMBER CHEMICAL NAME (POUNDS) (POUNDS) 1327-53-3 ARSENOUS OXIDE 100/500 1 7784-34-1 ARSENOUS TRICHLORIDE 500 1 7784-42-1 ARSINE 100 100 2642-71-9 AZINPHOS-ETHYL 100/500 100 86-50-0 AZINPHOS-METHYL 10/500 1 98-87-3 BENZAL CHLORIDE 500 5,000 98-16-8 BENZENAMINE, 3-(TRIFLUOROMETHYL)- 500 500 100-14-1 BENZENE, 1-(CHLOROMETHYL)-4-NITRO- 500/500 500 98-05-5 BENZENEARSONIC ACID 10/500 10 3615-21-2 BENZIMIDAZOLE, 4,5-DICHLORO-2-(TRI- 500/500 500 FLUOROMETHYL)- 98-07-7 BENZOTRICHLORIDE 100 10 100-44-7 BENZYL CHLORIDE 500 100 140-29-4 BENZYL CYANIDE 500 500 15271-41-7 BICYCLO[2.2.1]HEPTANE-2-CARBONITRILE,5- -
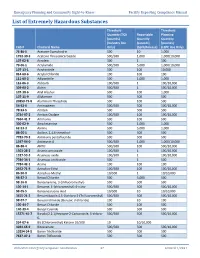
List of Extremely Hazardous Substances
Emergency Planning and Community Right-to-Know Facility Reporting Compliance Manual List of Extremely Hazardous Substances Threshold Threshold Quantity (TQ) Reportable Planning (pounds) Quantity Quantity (Industry Use (pounds) (pounds) CAS # Chemical Name Only) (Spill/Release) (LEPC Use Only) 75-86-5 Acetone Cyanohydrin 500 10 1,000 1752-30-3 Acetone Thiosemicarbazide 500/500 1,000 1,000/10,000 107-02-8 Acrolein 500 1 500 79-06-1 Acrylamide 500/500 5,000 1,000/10,000 107-13-1 Acrylonitrile 500 100 10,000 814-68-6 Acrylyl Chloride 100 100 100 111-69-3 Adiponitrile 500 1,000 1,000 116-06-3 Aldicarb 100/500 1 100/10,000 309-00-2 Aldrin 500/500 1 500/10,000 107-18-6 Allyl Alcohol 500 100 1,000 107-11-9 Allylamine 500 500 500 20859-73-8 Aluminum Phosphide 500 100 500 54-62-6 Aminopterin 500/500 500 500/10,000 78-53-5 Amiton 500 500 500 3734-97-2 Amiton Oxalate 100/500 100 100/10,000 7664-41-7 Ammonia 500 100 500 300-62-9 Amphetamine 500 1,000 1,000 62-53-3 Aniline 500 5,000 1,000 88-05-1 Aniline, 2,4,6-trimethyl- 500 500 500 7783-70-2 Antimony pentafluoride 500 500 500 1397-94-0 Antimycin A 500/500 1,000 1,000/10,000 86-88-4 ANTU 500/500 100 500/10,000 1303-28-2 Arsenic pentoxide 100/500 1 100/10,000 1327-53-3 Arsenous oxide 100/500 1 100/10,000 7784-34-1 Arsenous trichloride 500 1 500 7784-42-1 Arsine 100 100 100 2642-71-9 Azinphos-Ethyl 100/500 100 100/10,000 86-50-0 Azinphos-Methyl 10/500 1 10/10,000 98-87-3 Benzal Chloride 500 5,000 500 98-16-8 Benzenamine, 3-(trifluoromethyl)- 500 500 500 100-14-1 Benzene, 1-(chloromethyl)-4-nitro- 500/500 -

40 CFR Ch. I (7–1–11 Edition) Pt. 355, App. B
Pt. 355, App. B 40 CFR Ch. I (7–1–11 Edition) [Alphabetical Order] Reportable Threshold plan- CAS No. Chemical name Notes quantity * ning quantity (pounds) (pounds) 7550–45–0 ............ Titanium Tetrachloride ................................................ ..................... 1,000 100 584–84–9 .............. Toluene 2,4-Diisocyanate ........................................... ..................... 100 500 91–08–7 ................ Toluene 2,6-Diisocyanate ........................................... ..................... 100 100 110–57–6 .............. Trans-1,4-Dichlorobutene ........................................... ..................... 500 500 1031–47–6 ............ Triamiphos .................................................................. ..................... 500 500/10,000 24017–47–8 .......... Triazofos ..................................................................... ..................... 500 500 76–02–8 ................ Trichloroacetyl Chloride .............................................. ..................... 500 500 115–21–9 .............. Trichloroethylsilane ..................................................... d .................. 500 500 327–98–0 .............. Trichloronate ............................................................... e .................. 500 500 98–13–5 ................ Trichlorophenylsilane .................................................. d .................. 500 500 1558–25–4 ............ Trichloro(Chloromethyl)Silane .................................... ..................... 100 -

Environmental Protection Agency Pt. 355, App. A
Environmental Protection Agency Pt. 355, App. A agreement between a State and a sion over which the United States has Tribe, the SERC shall be the entity jurisdiction and Indian Country. identified in the agreement. Threshold planning quantity means, State means any State of the United for a substance listed in Appendices A States, the District of Columbia, the and B of this part, the quantity listed Commonwealth of Puerto Rico, Guam, in the column ‘‘threshold planning American Samoa, the United States quantity’’ for that substance. Virgin Islands, the Northern Mariana [73 FR 65462, Nov. 3, 2008, as amended at 73 Islands, any other territory or posses- FR 76960, Dec. 18, 2008] APPENDIX A TO PART 355—THE LIST OF EXTREMELY HAZARDOUS SUBSTANCES AND THEIR THRESHOLD PLANNING QUANTITIES [Alphabetical Order] Reportable Threshold plan- CAS No. Chemical name Notes quantity * ning quantity (pounds) (pounds) 75–86–5 ................ Acetone Cyanohydrin ................................................. ..................... 10 1,000 1752–30–3 ............ Acetone Thiosemicarbazide ....................................... ..................... 1,000 1,000/10,000 107–02–8 .............. Acrolein ....................................................................... ..................... 1 500 79–06–1 ................ Acrylamide .................................................................. f ................... 5,000 1,000/10,000 107–13–1 .............. Acrylonitrile ................................................................. f .................. -

Environmental Protection Agency Pt. 355, App. B
Environmental Protection Agency Pt. 355, App. B [Alphabetical Order] Reportable Threshold plan- CAS No. Chemical name Notes quantity * ning quantity (pounds) (pounds) 5344–82–1 ............ Thiourea, (2-Chlorophenyl)- ....................................... ..................... 100 100/10,000 614–78–8 .............. Thiourea, (2-Methylphenyl)- ....................................... ..................... 500 500/10,000 7550–45–0 ............ Titanium Tetrachloride ................................................ ..................... 1,000 100 584–84–9 .............. Toluene 2,4-Diisocyanate ........................................... ..................... 100 500 91–08–7 ................ Toluene 2,6-Diisocyanate ........................................... ..................... 100 100 110–57–6 .............. Trans-1,4-Dichlorobutene ........................................... ..................... 500 500 1031–47–6 ............ Triamiphos .................................................................. ..................... 500 500/10,000 24017–47–8 .......... Triazofos ..................................................................... ..................... 500 500 76–02–8 ................ Trichloroacetyl Chloride .............................................. ..................... 500 500 115–21–9 .............. Trichloroethylsilane ..................................................... d .................. 500 500 327–98–0 .............. Trichloronate ............................................................... e ................. -

Environmental Protection Agency Pt. 355, App. A
Environmental Protection Agency Pt. 355, App. A Release means any spilling, leaking, the facility is located. In the absence pumping, pouring, emitting, emptying, of a SERC for a State or Indian Tribe, discharging, injecting, escaping, leach- the Governor or the chief executive of- ing, dumping, or disposing into the en- ficer of the tribe, respectively, shall be vironment (including the abandonment the SERC. Where there is a cooperative or discarding of barrels, containers, agreement between a State and a and other closed receptacles) of any Tribe, the SERC shall be the entity hazardous chemical, EHS, or CERCLA identified in the agreement. hazardous substance. Solution means any aqueous or or- Reportable quantity means, for any ganic solutions, slurries, viscous solu- CERCLA hazardous substance, the tions, suspensions, emulsions, or quantity established in Table 302.4 of 40 pastes. CFR 302.4, for such substance. For any State means any State of the United EHS, reportable quantity means the States, the District of Columbia, the quantity established in Appendices A Commonwealth of Puerto Rico, Guam, and B of this part for such substance. American Samoa, the United States Unless and until superseded by regula- Virgin Islands, the Northern Mariana tions establishing a reportable quan- Islands, any other territory or posses- tity for newly listed EHSs or CERCLA sion over which the United States has hazardous substances, a weight of 1 jurisdiction and Indian Country. pound shall be the reportable quantity. Threshold planning quantity means, SERC means the State Emergency for a substance listed in Appendices A Response Commission for the State in and B of this part, the quantity listed which the facility is located except in the column ‘‘threshold planning where the facility is located in Indian quantity’’ for that substance. -

6 NYCRR Part
6 NYCRR PART 597 HAZARDOUS SUBSTANCES IDENTIFICATION, RELEASE PROHIBITION, AND RELEASE REPORTING (Statutory authority: Environmental Conservation Law sections 1-0101, 3-0301, 3-0303, 17-0301, 17-0303, 17-0501, 17-1743, 37-0101 through 37-0107, and 40-0101 through 40-0121) TABLE OF CONTENTS 597.1 General. ....................................................................................................................2 597.2 Criteria for identifying a hazardous substance or acutely hazardous substance. .....5 597.3 List of hazardous substances. ...................................................................................6 597.4 Spills and Releases of hazardous substances. ..........................................................6 Table 1 Hazardous Substances – Sorted by Name ................................................................9 Table 2 Hazardous Substances – Sorted by CAS Number .................................................49 6 NYCRR Part 597 – Oct. 11, 2015 Page 1 of 88 597.1 597.1 General. (a) Purpose. The purpose of this Part is to: (1) set forth criteria for identifying a hazardous substance or acutely hazardous substance; (2) set forth a list of hazardous substances; (3) identify reportable quantities for the spill or release of hazardous substances; (4) prohibit the unauthorized release of hazardous substances; and (5) establish requirements for reporting of releases of hazardous substances. (b) Definitions. The following definitions apply to this Part: (1) Authorized means the possession of a valid license, permit, or certificate issued by an agency of the state of New York or the federal government, or an order issued by the Department or United States Environmental Protection Agency under applicable statutes, rules or regulations regarding the possession or release of hazardous substances or otherwise engaging in conduct which is exempt under applicable statutes, rules or regulations from the requirements of possessing such a license, permit, certificate or order. -
Schafer, E. W., Jr., and W. A. Bowles Jr. 2004. Toxicity
United States Department of Agriculture Animal and Plant Health Toxicity, Repellency or Inspection Service Phytotoxicity of 979 Wildlife Services Chemicals to Birds, National Wildlife Research Center Mammals and Plants Research Report No. 04-01 The United States Department of Agriculture (USDA) prohibits discrimination in all its programs and activities on the basis of race, color, nat ional origin, gender, religion, age, disability, political beliefs, sexual orientation, or marital or family status. (Not all prohibited biases apply to all programs.) Persons with disabilities who require alternative means for communication of program information (Braille, large print, audiotape, etc.) should contact USDA’s TARGET Center at (202)720-2600 (voice and TDD). To file a complaint of discrimination, write USDA, Director, Office of Civil Rights, Room 326-W, Whitten Building, 14th and Independence Avenue, SW, Washington, DC 20250-9410 or call (202) 720-5964 (voice and TDD). USDA is an equal opportunity provider and employer. Mention of companies or commercial products does not imply recommendation or endorsement by USDA over others not mentioned. USDA neither guarantees nor warrants the standard of any product mentioned. Product names are mentioned solely to report factually on available data and to provide specific information. United States Department of Agriculture Animal and Plant Toxicity, Repellency Health Inspection Service or Phytotoxicity of Wildlife Services 979 Chemicals to Birds, National Wildlife Research Center Mammals and Plants Research Report No. 04-01 By: Edward W. Schafer, Jr. 1 Walter A. Bowles, Jr. 1 _ 1 Mr. Schafer and Mr. Bowles are retired, former employees of the USDA, APHIS, WS, NWRC, located in Fort Collins, Colorado. -

WO 2017/065602 Al 20 April 2017 (20.04.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/065602 Al 20 April 2017 (20.04.2017) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 31/365 (2006.01) A61P 25/28 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 31/475 (2006.01) A61K 45/06 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 31/50 (2006.01) A61K 31/43 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, A61K 31/5517 (2006.01) A61K 31/573 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/NL20 16/050490 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 7 July 2016 (07.07.2016) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 2015608 13 October 2015 (13. -

Appendix D State of California List of Regulated
California Refineries Outreach Programs anil Emergency Response Plans APPENDIX D STATE OF CALIFORNIA LIST OF REGULATED SUBSTANCES AND THRESHOLD QUANTITIES FOR ACCIDENTAL RELEASE PREVENTION Table 3. State Regulated Substances List and Threshold Quantities for Accidental Release Prevention Also on State Chemical Name Table 1' CAS Number Threshold Quantity (lbs) Acetone Cyanohydrin 2 no 75-86-5 1,000 Acetone Thiosemicarbazide no 1752-30-3 1,000/10,000 3 Acrolein yes 107-02-8 500 Acrylamide no 79-06-1 1,000/10,000 3 Acrylonitrile yes 107-13-1 10,000 Acrylyl Chloride yes 814-68-6 100 Aldicarb no t16-06-3 100/10,000 3 Aldrin no 309-00-2 500/10,000 3 Allyl Alcohol yes 107-18-6 1,000 Allyiamine yes 107-11-9 500 4 Aluminum Phosphide no 20859-73-8 500 Aminopterin no 54-62-6 500/10,000 3 Amiton Oxalate no 3734-97-2 100/10,000 3 AmmoniaA • 5 yes 7664-41-7 500 Aniline no 62-53-3 1,000 Antimycin A no 1397-94-0 1,000/10,000 3 ANTU no 86-88-4 500/10,000 3 Arsenic Pentoxide no 1303-28-2 100/10,000 3 Arsenous Oxide no 1327-53-3 100/10,000 3 Arsenous Trichloride yes . 7784-34-1 500 Arsine yes 7784-42-1 100 Azinphos-Ethyl no 2642-71-9 100/10,000 3 Azinphos-Methyl no 86-50-0 10/10,000 3 Benzene, l-(Chloromethyl)-4-Nitro- no 100-14-1 500/10,000 3 Benzenearsonic Acid no 98-05-5 10/10,000 3 Benzimidazole, 4,5-DichIoro-2-(Trifluoromethyl)- no 3615-21-2 500/10,000 3 Benzotrichloride 2 no 98-07-7 100 Bicyclo[2.2.1] Heptane-2-Carbonitrile, 5-Chloro- 6-((((Methyiamino) no 15271-41-7 500/10,000 3 Carbonyl)Oxy)Imino)', (1 s-( 1-alpha, 2-beta, 4-alpha, 5-alpha. -

Section 302 EHS TPQ List.Pdf
When two values are listed, the second A facility is subject to Emergency Planning under Section 302 of EPCRA (40 CFR Part value represents the 355) if any of the substances on this list are present at or above the Threshold Planning TPQ of the substance Quantity (TPQ) if present in solid form Section 302 (EHS) NAME TPQ 1,1-Dimethyl hydrazine 1,000 1,2-Ethanediamine 10,000 1,4:5,8-Dimethanonaphthalene, 1,2,3,4,10,10-hexachloro-1,4,4a,5,8,8a-hexahydro- (1.alpha.,4.alpha.,4a.beta.,5.alpha.,8.alpha.,8a.beta.)- 500/10,000 2,2'-Bioxirane 500 2,4-Dithiobiuret 100/10,000 2-Butenal 1,000 2-Butenal, (e)- 1,000 2-Chloro-N-(2-chloroethyl)-N-methylethanamine 10 2-Methyllactonitrile 1,000 2-Propen-1-amine 500 2-Propen-1-ol 1,000 2-Propenal 500 2-Propenenitrile 10,000 2-Propenenitrile, 2-methyl- 500 2-Propenoyl chloride 100 3-Chloropropionitrile 1,000 4,6-Dinitro-o-cresol 10/10,000 4,7-Methanoindan, 1,2,3,4,5,6,7,8,8-octachloro-2,3,3a,4,7,7a-hexahydro- 1,000 4-Aminopyridine 500/10,000 5-(Aminomethyl)-3-isoxazolol 500/10,000 5-Fluorouracil 500/10,000 Acetic acid ethenyl ester 1,000 Acetone cyanohydrin 1,000 Acetone thiosemicarbazide 1,000/10,000 Acrolein 500 Acrylamide 1,000/10,000 Acrylonitrile 10,000 Acrylyl chloride 100 Adiponitrile 1,000 Aldicarb 100/10,000 Aldrin 500/10,000 Allyl alcohol 1,000 Allylamine 500 Aluminum phosphide 500 Aminopterin 500/10,000 Amiton 500 Amiton oxalate 100/10,000 Ammonia 500 Ammonia (anhydrous) 500 Amphetamine 1,000 Aniline 1,000 Aniline, 2,4,6-trimethyl- 500 Antimony pentafluoride 500 Antimycin A 1,000/10,000 ANTU 500/10,000 -

SARA TITLE III EXTREMELY HAZARDOUS SUBSTANCES (Ehss)
SARA TITLE III EXTREMELY HAZARDOUS SUBSTANCES (EHSs) EHS NAME CAS NUMBER TPQ* 1,1-Dimethyl hydrazine 57-14-7 1,000 1,2-Ethanediamine 107-15-3 10,000 1,4:5,8-Dimethanonaphthalene, 1,2,3,4,10,10-hexachloro- 1,4,4a,5,8,8a-hexahydro- 309-00-2 500/10,000 (1.alpha.,4.alpha.,4a.beta.,5.alpha.,8.alpha.,8a.beta.)- 2,2'-Bioxirane 1464-53-5 500 2,4-Dithiobiuret 541-53-7 100/10,000 2-Butenal 4170-30-3 1,000 2-Butenal, (e)- 123-73-9 1,000 2-Chloro-N-(2-chloroethyl)-N-methylethanamine 51-75-2 10 2-Methyllactonitrile 75-86-5 1,000 2-Propen-1-amine 107-11-9 500 2-Propen-1-ol 107-18-6 1,000 2-Propenal 107-02-8 500 2-Propenenitrile 107-13-1 10,000 2-Propenenitrile, 2-methyl- 126-98-7 500 2-Propenoyl chloride 814-68-6 100 3-Chloropropionitrile 542-76-7 1,000 4,6-Dinitro-o-cresol 534-52-1 10/10,000 4,7-Methanoindan, 1,2,3,4,5,6,7,8,8-octachloro-2,3,3a,4,7,7a- 57-74-9 1,000 hexahydro- 4-Aminopyridine 504-24-5 500/10,000 5-(Aminomethyl)-3-isoxazolol 2763-96-4 500/10,000 5-Fluorouracil 51-21-8 500/10,000 Acetic acid ethenyl ester 108-05-4 1,000 Acetone cyanohydrin 75-86-5 1,000 Acetone thiosemicarbazide 1752-30-3 1,000/10,000 Acrolein 107-02-8 500 Acrylamide 79-06-1 1,000/10,000 Acrylonitrile 107-13-1 10,000 Acrylyl chloride 814-68-6 100 Adiponitrile 111-69-3 1,000 Aldicarb 116-06-3 100/10,000 Aldrin 309-00-2 500/10,000 Allyl alcohol 107-18-6 1,000 Allylamine 107-11-9 500 Aluminum phosphide 20859-73-8 500 Aminopterin 54-62-6 500/10,000 Amiton 78-53-5 500 Amiton oxalate 3734-97-2 100/10,000 Ammonia 7664-41-7 500 Ammonia (anhydrous) 7664-41-7 500 Amphetamine 300-62-9 1,000 Aniline 62-53-3 1,000 Aniline, 2,4,6-trimethyl- 88-05-1 500 Antimony pentafluoride 7783-70-2 500 Antimycin A 1397-94-0 1,000/10,000 ANTU 86-88-4 500/10,000 Arsenic pentoxide 1303-28-2 100/10,000 *TPQ = Threshold Planning Quanitity in pounds.