In Acidic Chloride Brines Under Hydrothermal Conditions: from Transport to Deposition of Uranium in Unconformity-Related Deposits Maxime Dargent
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gasket Chemical Services Guide
Gasket Chemical Services Guide Revision: GSG-100 6490 Rev.(AA) • The information contained herein is general in nature and recommendations are valid only for Victaulic compounds. • Gasket compatibility is dependent upon a number of factors. Suitability for a particular application must be determined by a competent individual familiar with system-specific conditions. • Victaulic offers no warranties, expressed or implied, of a product in any application. Contact your Victaulic sales representative to ensure the best gasket is selected for a particular service. Failure to follow these instructions could cause system failure, resulting in serious personal injury and property damage. Rating Code Key 1 Most Applications 2 Limited Applications 3 Restricted Applications (Nitrile) (EPDM) Grade E (Silicone) GRADE L GRADE T GRADE A GRADE V GRADE O GRADE M (Neoprene) GRADE M2 --- Insufficient Data (White Nitrile) GRADE CHP-2 (Epichlorohydrin) (Fluoroelastomer) (Fluoroelastomer) (Halogenated Butyl) (Hydrogenated Nitrile) Chemical GRADE ST / H Abietic Acid --- --- --- --- --- --- --- --- --- --- Acetaldehyde 2 3 3 3 3 --- --- 2 --- 3 Acetamide 1 1 1 1 2 --- --- 2 --- 3 Acetanilide 1 3 3 3 1 --- --- 2 --- 3 Acetic Acid, 30% 1 2 2 2 1 --- 2 1 2 3 Acetic Acid, 5% 1 2 2 2 1 --- 2 1 1 3 Acetic Acid, Glacial 1 3 3 3 3 --- 3 2 3 3 Acetic Acid, Hot, High Pressure 3 3 3 3 3 --- 3 3 3 3 Acetic Anhydride 2 3 3 3 2 --- 3 3 --- 3 Acetoacetic Acid 1 3 3 3 1 --- --- 2 --- 3 Acetone 1 3 3 3 3 --- 3 3 3 3 Acetone Cyanohydrin 1 3 3 3 1 --- --- 2 --- 3 Acetonitrile 1 3 3 3 1 --- --- --- --- 3 Acetophenetidine 3 2 2 2 3 --- --- --- --- 1 Acetophenone 1 3 3 3 3 --- 3 3 --- 3 Acetotoluidide 3 2 2 2 3 --- --- --- --- 1 Acetyl Acetone 1 3 3 3 3 --- 3 3 --- 3 The data and recommendations presented are based upon the best information available resulting from a combination of Victaulic's field experience, laboratory testing and recommendations supplied by prime producers of basic copolymer materials. -
NOI to Prepare an Environmental Impact Statement For
Federal Register / Vol. 64, No. 34 / Monday, February 22, 1999 / Notices 8553 reauthorization of OERI and will hear DEPARTMENT OF ENERGY Orders are summarized in the attached committee and officers' reports. A final appendix. [FE Docket Nos. 99±01±NG, 99±03±NG, 99± agenda will be available from the Board 02±NG, 99±04±NG, 92±24±NG, 99±05±NG, These Orders may be found on the FE office on March 10, 1999, and will be and 99±06±NG] web site at http://www.fe.doe.gov., or posted on the Board's web site, http:// on the electronic bulletin board at (202) www.ed.gov/offices/OERI/NERPPB/. Office of Fossil Energy; Orders 586±7853. Granting and Transferring Records are kept of all Board They are also available for inspection Authorizations To Import and/or Export proceedings and are available for public and copying in the Office of Natural Gas Natural Gas inspection at the office of the National & Petroleum Import & Export Activities, Educational Research Policy and OGE Energy Resources, Inc., National Fuel Docket Room 3E±033, Forrestal Priorities Board, Suite 100, 80 F St., Gas Distribution Corporation, Renaissance Building, 1000 Independence Avenue, NW, Washington, D.C. 20208±7564. Energy (U.S.) Inc., Selkirk Cogen Partners, SW, Washington, DC 20585, (202) 586± L.P., Coral Energy Resources, L.P. (Successor 9478. The Docket Room is open between Dated: February 16, 1999. to Salmon Resources Ltd.), Transco Energy the hours of 8:00 a.m. and 4:30 p.m., Eve M. Bither, Marketing Company, and Petro-Canada Hydrocarbons Inc. -

This Generic Chemical Compatibility Chart Is Offered Only As a General
Chemical Compatibility Chart This Generic Chemical Compatibility Chart is offered only as a general guide, indicating the suitability of various elastomers for service in these chemicals and fluids. The ratings are based, for the most part, on published literature of various polymer suppliers, rubber manufacturers, and in some cases, the considered opinion of experienced compounders. We cannot guarantee their accuracy nor assume any responsibility for use thereof. Therefore, we recommend that you always TEST your materials for your particular application before moving to production. www.GlobalORing.com 4250 N. Sam Houston Parkway E, Suite 100, Houston, TX 77032 Phone: 832-448-5550 / 877-448-5550 Fax: 832-448-5551 / 877-448-5551 Chemical Compatibility Chart Polymer Comparison Temperature Durometer Media Resistance ASTM Range (°F) Range Abbr Low High Low High Oil Fuel Acid Alkali Ozone Steam Brake Weather ACM -40 400 50 80 E P – G P -F G - E G - E P P E AEM -50 350 40 90 F P – G P – F P – F E P P E AU -65 250 40 90 G P – G P – G P – F E P P E CIIR -50 300 60 70 P P F – E G - E E G – E G F CM -60 300 80 80 G – E P – F G – E E E P - F G E CR -80 220 30 90 F – G P – F F – G G – P G – E P – G F P - G CSM -65 275 50 80 E F G – E G – E E – O P – G F E – O ECO -80 to -15 275 to 325 50 90 E G - E P - G P – G G – E F – G P G – E EPDM -75 350 40 90 P P G G G - E E G – E E EU -65 250 40 90 G P – G P – G P – F E P P E FEPM 0 450 to 500 70 90 E P - F F - G E E G - E F – E E FKM -20 to 20 500 50 90 E E G - E P - G O P – G F E FVMQ -110 to -90 450 40 -

EPDM & FKM Chemical Resistance Guide
EPDM & FKM Chemical Resistance Guide FIRST EDITION EPDM & FKM CHEMICAL RESISTANCE GUIDE Elastomers: Ethylene Propylene (EPDM) Fluorocarbon (FKM) Chemical Resistance Guide Ethylene Propylene (EPDM) & Fluorocarbon (FKM) 1st Edition © 2019 by IPEX. All rights reserved. No part of this book may be used or reproduced in any manner whatsoever without prior written permission. For information contact: IPEX, Marketing, 1425 North Service Road East, Oakville, Ontario, Canada, L6H 1A7 ABOUT IPEX At IPEX, we have been manufacturing non-metallic pipe and fittings since 1951. We formulate our own compounds and maintain strict quality control during production. Our products are made available for customers thanks to a network of regional stocking locations from coast-to-coast. We offer a wide variety of systems including complete lines of piping, fittings, valves and custom-fabricated items. More importantly, we are committed to meeting our customers’ needs. As a leader in the plastic piping industry, IPEX continually develops new products, modernizes manufacturing facilities and acquires innovative process technology. In addition, our staff take pride in their work, making available to customers their extensive thermoplastic knowledge and field experience. IPEX personnel are committed to improving the safety, reliability and performance of thermoplastic materials. We are involved in several standards committees and are members of and/or comply with the organizations listed on this page. For specific details about any IPEX product, contact our customer service department. INTRODUCTION Elastomers have outstanding resistance to a wide range of chemical reagents. Selecting the correct elastomer for an application will depend on the chemical resistance, temperature and mechanical properties needed. Resistance is a function both of temperatures and concentration, and there are many reagents which can be handled for limited temperature ranges and concentrations. -
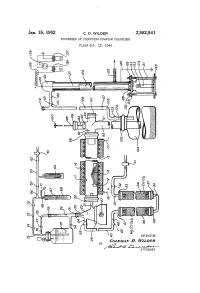
Jan. 15, 1952 C. D., WLDER 2,582,941 PROCESSES of PRODUCING URANIUM CHLORIDES Filed Oct
Jan. 15, 1952 C. D., WLDER 2,582,941 PROCESSES OF PRODUCING URANIUM CHLORIDES Filed Oct. 2, 1944 d S Q Q Q t 2. INVENTOR. 6ozEMAW Z. WZZZOER ATTORNEY . Patented Jan. 15, 1952 2,582,941 UNITED STATES PATENT OFFICE 2,582,941 PROCESSES OF PRODUCING URANUM CHELORDES - Coleman D. Wilder, Oak Ridge, Tenn., assignor to the United States. of America, as represented bysion the United States Atomic Energy Commis Application October 12, 1944, Serial No. 558,452 8 Claims. (C1. 23-145) 2. - This invention relates to the manufacture of a pating carbon tetrachloride vapor in a speedy uranium chloride product, and more particularly stream of dry air, passing the resultant mixture re of uranium pentachloride over an uranium compound heated to the neigh - - - - - a . g a substantial proportion of borhood of 550° C. whereby uranium pentachlo uranium hexachloride, by a process for chlorinat: ride is formed and vaporized into the current, ing various, compositions comprising uranium moving the resulting vaporous mixture into a compounds, with carbon tetrachloride vapor car collecting receptacle so that most of the uranium ried by a Swift current of air. chloride will condense and settle out, and send This invention has for an object the rapid pro ing the gas stream through a dust separator to duction of a uranium chloride product compris strip it of the residual uranium-containing par ing a large proportion of uranium pentachloride ticles. and various proportions of uranium hexachlo The apparatus employed comprises a device ride. for supplying carbon tetrachloride, a device for A further object of the invention is to provide supplying dry air, a flash boiler in which the a high-yield, low-loss process for the production 5 carbon tetrachloride is vaporized and mixed with of uranium pentachloride which is economical the air, a reaction chamber in which the ai and suitable for large scale production of said carbon tetrachloride admixture is reacted with chloride. -
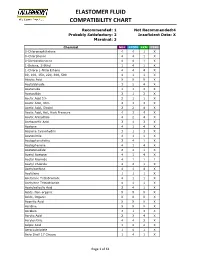
Elastomer Fluid Compatibility Chart
ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE 0-Chloronaphthalene 4 4 1 X 0-Chlorphenol 4 4 1 X 0-Dichlorobenzene 4 4 1 X 1-Butene, 2-Ethyl 1 4 1 X 1-Chloro 1-Nitro Ethane 4 4 4 X 90, 100, 150, 220, 300, 500 4 1 1 X Abietic Acid X X X X Acetaldehyde 3 2 4 X Acetamide 1 1 3 X Acetanilide 3 1 3 X Acetic Acid 5% 2 1 1 X Acetic Acid, 30% 2 1 3 X Acetic Acid, Glacial 2 2 4 X Acetic Acid, Hot, High Pressure 4 3 4 X Acetic Anhydride 4 2 4 X Acetoacetic Acid 3 1 3 X Acetone 4 1 4 X Acetone Cyanohydrin 3 1 3 X Acetonitrile 3 1 1 X Acetophenetidine 2 4 1 X Acetophenone 4 1 4 X Acetotoluidide 2 4 1 X Acetyl Acetone 4 1 4 X Acetyl Bromide 4 1 1 1 Acetyl Chloride 4 4 1 X Acetylacetone 4 1 4 X Acetylene 1 1 1 X Acetylene Tetrabromide 4 1 1 X Acetylene Tetrachloride 4 1 1 X Acetylsalicylic Acid 2 4 1 X Acids, Non-organic X X X X Acids, Organic X X X X Aconitic Acid X X X X Acridine X X X X Acrolein 3 1 3 X Acrylic Acid 2 3 4 X Acrylonitrile 4 4 3 X Adipic Acid 1 2 2 X Aero Lubriplate 1 4 1 X Aero Shell 17 Grease 1 4 1 X Page 1 of 61 ELASTOMER FLUID COMPATIBILITY CHART Recommended: 1 Not Recommended:4 Probably Satisfactory: 2 Insuficient Data: X Marginal: 3 Chemical NBR EPDM FKM PTFE Aero Shell 1AC Grease 1 4 1 X Aero Shell 750 2 4 1 X Aero Shell 7A Grease 1 4 1 X Aerosafe 2300 4 1 4 X Aerosafe 2300W 4 1 4 X Aerozene 50 (50% Hydrazine 50% UDMH) 3 1 4 X Air, 1 1 1 X Air, 200 - 300° F 3 2 1 X Air, 300 - 400° F 4 4 1 X Air, 400 - 500° F 4 4 3 X Air, -

Fission Yield Measurements from Deuterium-Tritium Fusion Produced Neutrons Using Cyclic Neutron Activation Analysis and Γ-Γ Coincidence Counting
Fission yield measurements from deuterium-tritium fusion produced neutrons using cyclic neutron activation analysis and γ-γ coincidence counting by Bruce D. Pierson A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Nuclear Engineering & Radiological Sciences) in The University of Michigan 2016 Doctoral Committee: Professor Sara A. Pozzi, co-chair Assistant Professor Marek Flaska, Penn. State University, co-chair Professor, John E. Foster Larry R. Greenwood, Pacific Northwest National Laboratory Assistant Professor Physics Thomas Schwarz c Bruce D. Pierson 2016 All Rights Reserved This dissertation is dedicated to my family for their unyielding patience and support throughout my graduate career. ii ACKNOWLEDGEMENTS I would like to thank Drs. Marek Flaska, Larry Greenwood, Amanda Prinke, Sara Pozzi, and Sean Stave for their assistance, guidance, mentorship, and revisions to written works; their support and input drastically improved the quality of the final analysis and results (between the five them, I was getting at least one form of support from each of them). I'd also like to thank Drs. Ovidiu Toader and Joe Miklos for their assistance and support in maintaining and managing the Neutron Science Laboratory. Dr. Miklos was instrumental in amending the Nuclear Science Laboratory Nuclear Regulatory Commission license that allowed me to even do the work outlined in this document. He is a good friend and cheered me on to the finish at every opportunity. Dr. Toader was an invaluable resource for tools and ideas, and even emotional support when confronted with complex problems and the, what seemed to be, near endless graduate career. -

Lawrence Berkeley National Laboratory Recent Work
Lawrence Berkeley National Laboratory Recent Work Title THE KINETICS OF THE CHLORINATION OF UC14 BY Cl2 Permalink https://escholarship.org/uc/item/28x84687 Author Camahort, Jose Luis Publication Date 1965-05-20 eScholarship.org Powered by the California Digital Library University of California UCRL-11986 University of California Ernest 0. lawrence Radiation Laboratory THE KINETICS OF THE CHLORINATION OF UC1 BY C12 - TWO-WEEK LOAN COPY This is a library Circulating Copy which may be borrowed for two weeks. For a personal retention copy, call Tech. Info. Dioision, Ext. 5545 Berkeley, California DISCLAIMER This document was prepared as an account of work sponsored by the United States Government. While this document is believed to contain correct information, neither the United States Government nor any agency thereof, nor the Regents of the University of California, nor any of their employees, makes any warranty, express or implied, or assumes any legal responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by its trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof, or the Regents of the University of California. The views and opinions of authors expressed herein do not necessarily state or reflect those of the United States Government or any agency thereof or the Regents of the University of California. DCl\L-llSJ36 UNIVERSITY OF CALIFORNIA Lal·rrence Radiation Laboratory Berkeley} California AEC Contract No. -

2,546,700 March 27, 1951
March 27, 1951 PRESCOTT, JR MULTIPLE CHISEL STRUCTURE FOR DISINTEGRATING AND REMOWING2,546,700 INCRUSTATIONS FROM INTERIOR WALLS OF RECEPTACLES Original Filled Sept. 2, 1944 2. Sheets-Sheet INVENTOR. 6A74azas/7 A2asco 77/2, 2 ATTORNEY. March 27, 1951 c. H. PREscoTT, JR 2,546,700 MULTIPLE CHISEL STRUCTURE FOR DISINTEGRATING AND REMOWING INCRUSTATIONS FROM INTERIOR WALLS OF RECEPTACLES Original Filled Sept. 2, l944 2. Sheets-Sheet 2 -/03 136 2 2 A?/ x-42W12 E -54 S a3 A76.5 S. 3 2 R R 2 /46 /44 /74 /64 /4O s% 3 s &a /42, 2:/72 Š&24ze N i S s 76 f 5.s s s 3:y a NN SN 24/32 %io Ave. 7 INVENTOR. Gaezais /7. A2a56O77. /e. BY ATTORNEY. Patented Mar. 27, 1951 2,546,700 UNITED STATES PATENT OFFICE 2,546,700 MULTIPLE CHISEL STRUCTURE FOR DIS INTEGRATING AND REMOVING INCRUS TATIONS FROM INTERIOR, WALLS OF RECEPTACLES Charles H. Prescott, Jr., Berkeley, Calif., assignor to the United States of America as represented by the United States Atomic Energy Commis sion Original application September 2, 1944, Serial No. 552,556. Divided and this application Novem ber 2, 1945, Serial No. 626,388 4 Claims. (CI, 15-104.16) This invention relates to apparatus for use in aging the hygroscopic uranium compound com connection with packaging and/or closing, in the prises a subliming chamber, a condensing cham absence of air, receptacles containing material ber joined thereto, a storage receptacle and a that is rapidly decomposed or otherwise affected conduit connecting the condensing chamber and deleteriously when exposed to atmospheric con storage receptacle, means for maintaining a vacu ditions. -

LCSH Section U
U-2 (Reconnaissance aircraft) (Not Subd Geog) U.S. 29 U.S. Bank Stadium (Minneapolis, Minn.) [TL686.L (Manufacture)] USE United States Highway 29 BT Stadiums—Minnesota [UG1242.R4 (Military aeronautics)] U.S. 30 U.S. Bicycle Route System (May Subd Geog) UF Lockheed U-2 (Airplane) USE United States Highway 30 UF USBRS (U.S. Bicycle Route System) BT Lockheed aircraft U.S. 31 BT Bicycle trails—United States Reconnaissance aircraft USE United States Highway 31 U.S.-Canada Border Region U-2 (Training plane) U.S. 40 USE Canadian-American Border Region USE Polikarpov U-2 (Training plane) USE United States Highway 40 U.S. Capitol (Washington, D.C.) U-2 Incident, 1960 U.S. 41 USE United States Capitol (Washington, D.C.) BT Military intelligence USE United States Highway 41 U.S. Capitol Complex (Washington, D.C.) Military reconnaissance U.S. 44 USE United States Capitol Complex (Washington, U-Bahn-Station Kröpcke (Hannover, Germany) USE United States Highway 44 D.C.) USE U-Bahnhof Kröpcke (Hannover, Germany) U.S. 50 U.S. Cleveland Post Office Building (Punta Gorda, Fla.) U-Bahnhof Kröpcke (Hannover, Germany) USE United States Highway 50 UF Cleveland Post Office Building (Punta Gorda, UF Kröpcke, U-Bahnhof (Hannover, Germany) U.S. 51 Fla.) Station Kröpcke (Hannover, Germany) USE United States Highway 51 BT Post office buildings—Florida U-Bahn-Station Kröpcke (Hannover, Germany) U.S. 52 U.S. Coast Guard Light Station (Jupiter Inlet, Fla.) BT Subway stations—Germany USE United States Highway 52 USE Jupiter Inlet Light (Fla.) U-Bahnhof Lohring (Bochum, Germany) U.S. -

Toxicological Profile for Uranium
URANIUM 39 3. HEALTH EFFECTS 3.1 INTRODUCTION The primary purpose of this chapter is to provide public health officials, physicians, toxicologists, and other interested individuals and groups with an overall perspective on the toxicology of uranium. It contains descriptions and evaluations of toxicological studies and epidemiological investigations and provides conclusions, where possible, on the relevance of toxicity and toxicokinetic data to public health. A glossary and list of acronyms, abbreviations, and symbols can be found at the end of this profile. The health effects associated with oral or dermal exposure to natural and depleted uranium appear to be primarily chemical in nature and not radiological, while those from inhalation exposure may also include a slight radiological component, especially if the exposure involves prolonged exposure to insoluble uranium compounds. This profile is primarily concerned with the effects of exposure to natural and depleted uranium, but does include limited discussion regarding enriched uranium, which is considered to be more of a radiological than a chemical hazard. Also, whenever the term Aradiation@ is used, it applies to ionizing radiation and not to non-ionizing radiation. Although natural and depleted uranium are primarily chemical hazards, the next several paragraphs describe the radiological nature of the toxicologically-important uranium isotopes, because individual isotopes are addressed in some of the health effects studies. Uranium is a naturally occurring radioactive element and a member of the actinide series. Radioactive elements are those that undergo spontaneous transformation (decay), in which energy is released (emitted) either in the form of particles, such as alpha or beta particles, or electromagnetic radiation with energies sufficient to cause ionization, such as gamma rays or x-rays. -
Á4-2-4-2-2-121Attorney
Oct. 23, 1951 F. A. JENKNS 2,572,156 PROCESS OF PRODUCING URANIUM HEXACHLORIDE Filed July 13, 1943 12 2/ To Vacuum Line // Original Charge | Residue UCls INVENTOR. Francis A. Jenkins -á4-2-4-2-2-121ATTORNEY. Patented Oct. 23, 1951 2,572,156 UNITED STATES PATENT of FICE PROCESSES OF PRODUCING URANUM HEXACHORDE Francis A. Jenkins, Berkeley, Calif., assignor to the United States of America as represented by the United States Atomic Energy Commission Application July 13, 1943, Serial No. 494,447 Claims. (CI.23-14.5) 1 The present invention relates to UCls and suitable charge of uranium pentachloride is processes of producing the Same. placed in a substantially conventional molecular Heretofore the compound UCls has not been Still which comprises a hot Surface heated in any known. Moreover, it has been predicted that the Suitable manner, such, for example, as by a hot possibility of its production is virtually negligible Sandbath, and a cold Surface. cooled in any Suit in view of its expected great instability. This able manner, Such, for example, as by liquid air. view is entirely plausible since the tetrahalides In the still the hot surface and the cold surface of uranium form a series decreasing in stability are disposed in Spaced apart relation, and the in a regular-manner-from the fluoride through space therebetween is evacuated to -an-extremely the chloride-and-the-bromide to the iodide, and O low pressure by Suitable vacuum pumping ap since UF6 is... extremely unstable. Thus it has paratus. More particularly, the charge is placed always been assumed that the hexahalide Series on the hot surface of the still and the tempera of uranium terminates abruptly at the COm ture of the sand bath is suitably controlled, in pound UEs, .