The Mechanism Op Permanganate Oxidations
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Carbonyl Compounds
Carbonyl Compounds What are Carbonyl Compounds? Carbonyl compounds are compounds that contain the C=O (carbonyl) group. Preparation of Aldehydes: 1. Preparation from Acid Chloride (Rosenmund Reduction): This reaction was named after Karl Wilhelm Rosenmund, who first reported it in 1918. The reaction is a hydrogenation process in which an acyl chloride is selectively reduced to an aldehyde. The reaction, a hydrogenolysis, is catalysed by palladium on barium sulfate, which is sometimes called the Rosenmund catalyst. 2. Preparation from Nitriles: This reaction involves the preparation of aldehydes (R-CHO) from nitriles (R- CN) using SnCl2 and HCl and quenching the resulting iminium salt ([R- + − CH=NH2] Cl ) with water (H2O). During the synthesis, ammonium chloride is also produced. The reaction is known as Stephen Aldehyde synthesis. Dr. Sumi Ganguly Page 1 3. Preparation from Grignard Reagent: When Grignard Reagent is reacted with HCN followed by hydrolysis aldehyde is produced. Preparation of Ketones: 1. Preparation from Acid Chloride (Friedel-Crafts Acylation): Acid chlorides when reacted with benzene in presence of anhydrous AlCl3, aromatic ketone are produced. However, only aromatic ketones can be prepared by following this method. In order to prepare both aromatic and aliphatic ketones acid chlorides is reacted with lithium dialkylcuprate (Gilman Reagnt). Dr. Sumi Ganguly Page 2 The lithium dialkyl cuprate is produced by the reaction of two equivalents of the organolithium reagent with copper (I) iodide. Example: 3. Preparation from Nitriles and Grignard Reagents: When Grignard Reagent is reacted with RCN followed by hydrolysis aldehyde is produced. Dr. Sumi Ganguly Page 3 Physical Characteristic of Carbonyl Compounds: 1) The boiling point of carbonyl compounds is higher than the alkanes with similar Mr. -

Decolorization of Reactive Orange 16 Via Ferrate(VI) Oxidation Assisted by Sonication
Turkish Journal of Chemistry Turk J Chem (2017) 41: 577 { 586 http://journals.tubitak.gov.tr/chem/ ⃝c TUB¨ ITAK_ Research Article doi:10.3906/kim-1701-8 Decolorization of reactive orange 16 via ferrate(VI) oxidation assisted by sonication Serkan S¸AHINKAYA_ ∗ Department of Environmental Engineering, Faculty of Engineering and Architecture, Nev¸sehirHacı Bekta¸sVeli University, Nev¸sehir,Turkey Received: 02.01.2017 • Accepted/Published Online: 24.03.2017 • Final Version: 05.09.2017 Abstract: The decolorization of azo dye C.I. reactive orange 16 (RO 16) via ferrate(VI) and sono-ferrate(VI) methods, which is the combination of the ferrate(VI) oxidation method with sonication, has been achieved in the present study. The influences of some important operating parameters, which are the initial pH, the concentration of potassium ferrate(VI) (K 2 FeO 4) and the RO 16 dye, and ultrasonic density (for only the sono-ferrate(VI) method), on the color removal have −1 been investigated. The optimum conditions have been determined as pH = 7 and [K 2 FeO 4 ] = 50 mg L for the −1 −1 individual ferrate(VI) oxidation method and pH = 7 and [K 2 FeO 4 ] = 50 mg L by direct sonication at 0.50 W mL ultrasonic density and 20 kHz fixed frequency for the sono-ferrate(VI) method. The color removal efficiencies were 85% by ferrate(VI) method and 91% by sono-ferrate(VI) method. Kinetic studies were also performed for the decolorization of RO 16 under the optimized conditions at room temperature. It was seen that the oxidative decolorization of RO 16 via the sono-ferrate(VI) method happened more rapidly because of the production of OH • radical through sonication compared to the individual ferrate(VI) method. -

Kinetic Studies on Permanganate Oxidation of Acetophenones Under
Indi an Journal of Chemistry Vol. 40A, June 2001, pp. 610-612 Kinetic studies on permanganate oxidation of The ketones were further purified by vacuum acetophenones under phase transfer catalysis distillation. The solvents employed were purified by standard methods and doubly distilled water was P S Sheeba & T D Radhakrishnan Nair* always used. The catalysts used were tricapryl Department of Chemistry, Calicut University methylammonium chloride (TCMAC) and tetrabutyl Calicut University P.O., Kerala 673 635. India ammonium bromide (TBAB). The former has Received 18 October 2000; revised 8 March 2001 relatively larger organic structure (C 10H21 )JN+CH 3Cr compared to the latter (C4 H9) 4N+B(. Kinetic studies on the permanganate oxidation of The solutions containing the permanganate ions in acetopheno_ne and some of its substituents in organic media using the organic solvents were prepared by shaking the techn1que of phase transfer catalysis are reported. aqueous potassium permanganate solution with the Tncaprylmethylammonium chloride and tetrabutylammonium bromide have been used as the phase transfer catalysts. The organic solvents contammg the phase transfer 4 reaction shows first order dependence each on [ketones] and the catalysts as reported elsewhere . The solutions of the [permanganate ions] respectively . The rate coefficients fit well oxidant thus prepared in the organic solvent and the with the Hammett equation and the p-value calculated aorees well substrate in the organic solvent were thermally with the reaction requirements. "' equilibrated (± 0.05°C) at the desired temperature for Potassium permanganate is a powerful oxidizing about half an hour before mixing. Kinetic runs were carried out under pseudo-first order conditions. -

Manufacturing of Potassium Permanganate Kmno4 This Is the Most Important and Well Known Salt of Permanganic Acid
Manufacturing of Potassium Permanganate KMnO4 This is the most important and well known salt of permanganic acid. It is prepared from the pyrolusite ore. It is prepared by fusing pyrolusite ore either with KOH or K2CO3 in presence of atmospheric oxygen or any other oxidising agent such as KNO3. The mass turns green with the formation of potassium manganate, K2MnO4. 2MnO2 + 4KOH + O2 →2K2MnO4 + 2H2O 2MnO2 + 2K2CO3 + O2 →2K2MnO4 + 2CO2 The fused mass is extracted with water. The solution is now treated with a current of chlorine or ozone or carbon dioxide to convert manganate into permanganate. 2K2MnO4 + Cl2 → 2KMnO4 + 2KCl 2K2MnO4 + H2O + O3 → 2KMnO4 + 2KOH + O2 3K2MnO4 + 2CO2 → 2KMnO4 + MnO2 + 2K2CO3 Now-a-days, the conversion is done electrolytically. It is electrolysed between iron cathode and nickel anode. Dilute alkali solution is taken in the cathodic compartment and potassium manganate solution is taken in the anodic compartment. Both the compartments are separated by a diaphragm. On passing current, the oxygen evolved at anode oxidises manganate into permanganate. At anode: 2K2MnO4 + H2O + O → 2KMnO4 + 2KOH 2- - - MnO4 → MnO4 + e + - At cathode: 2H + 2e → H2 Properties: It is purple coloured crystalline compound. It is fairly soluble in water. When heated alone or with an alkali, it decomposes evolving oxygen. 2KMnO4 → K2MnO4 + MnO2 + O2 4KMnO4 + 4KOH → 4K2MnO4 + 2H2O + O2 On treatment with conc. H2SO4, it forms manganese heptoxide via permanganyl sulphate which decomposes explosively on heating. 2KMnO4+3H2SO4 → 2KHSO4 + (MnO3)2SO4 + 2H2O (MnO3)2SO4 + H2O → Mn2O7 + H2SO4 Mn2O7 → 2MnO2 + 3/2O2 Potassium permanganate is a powerful oxidising agent. A mixture of sulphur, charcoal and KMnO4 forms an explosive powder. -

Revision Guide
Revision Guide Chemistry - Unit 3 Physical and Inorganic Chemistry GCE A Level WJEC These notes have been authored by experienced teachers and are provided as support to students revising for their GCE A level exams. Though the resources are comprehensive, they may not cover every aspect of the specification and do not represent the depth of knowledge required for each unit of work. 1 Content Page Section 2 3.1 – Redox and standard electrode potential 13 3.2 - Redox reactions 20 3.3 - Chemistry of the p-block 30 3.4 - Chemistry of the d-block transition metals 35 3.5 - Chemical kinetics 44 3.6 - Enthalpy changes for solids and solutions 50 3.7 - Entropy and feasibility of reactions 53 3.8 - Equilibrium constants 57 3.9 - Acid-base equilibria 66 Acknowledgements 2 3.1 – Redox and standard electrode potential Redox reactions In AS, we saw that in redox reactions, something is oxidised and something else is reduced (remember OILRIG – this deals with loss and gain of electrons). Another way that we can determine if a redox reaction has happened is by using oxidation states or numbers (see AS revision guide pages 2 and 44). You need to know that: - • oxidation is loss of electrons • reduction is gain of electrons • an oxidising agent is a species that accepts electrons, thereby helping oxidation. It becomes reduced itself in the process. • a reducing agent is a species that donates electrons, thereby helping reduction. It becomes oxidised itself in the process. You also should remember these rules for assigning oxidation numbers in a compound: - 1 All elements have an oxidation number of zero (including diatomic molecules like H2) 2 Hydrogen is 1 unless it’s with a Group 1 metal, then it’s -1 3 Oxygen is -2 (unless it’s a peroxide when it’s -1, or reacted with fluorine, when it’s +2). -

Experiment: Determination of Manganese in Steel
p.1 of 4 Chemistry 201 – Winter 2007 Experiment: Determination of Manganese in Steel Manganese (Mn) in steel may be determined upon dissolution as manganese (VII) after oxidation from the manganese oxidation state (II). This procedure calls for three oxidizing agents. Manganese (VII) may be determined spectrophotometrically. During dissolution of the sample with dilute nitric acid to form iron (II) and manganese (II), nitrogen oxide gases are formed. These must be removed, partially by boiling, and the rest with ammonium peroxydisulfate which also removes carbon or other organic matter present. The nitrogen oxides may react with periodic acid which is used to convert manganese to the (VII) oxidation state. Excess peroxydisulfate is decomposed by boiling. The equations are: - + 2+ 3 Mn + 2 NO3 + 8H ---> 3 Mn + 2 NO (g) + 4H2O 2- - 2- + 2 NO2 (g) + S2O8 + 2 H2O ---> 2 NO3 + 2 SO4 + 4 H 2- 2- + 2 S2O8 + 2 H2O + heat ---> 4 SO4 + O2 + 4 H 2+ - - + - 2 Mn + 5 IO4 + 3 H2O ---> 2 MnO4 + 6 H + 5 IO3 NO(colorless gas) + 1/2 O2 (g) <---> NO2 (brown gas) Note: Peroxydisulfate has the required oxidizing potential to change the oxidation state of manganese from (II) to (IV), but the reaction is too slow. Silver could be employed catalytically but the results are too erratic. Periodate oxidizes the manganese to the (VII) state quantitatively and rapidly at the boiling point, and it is advisable to add the sparingly soluble periodate salt in several portions to maintain an excess and to allow for some decomposition of the latter salt at the elevated temperatures. -
A Manganate Ester Hydrolysis Mn(IV)
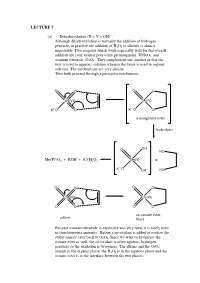
LECTURE 7 (a) Dihydroxylation (X = Y = OH) Although dihydroxylation is formally the addition of hydrogen peroxide, in practice the addition of H2O2 to alkenes is almost impossible. Two reagents which work especially well for this overall addition are cold, neutral potassium permanganate, KMnO4, and osmium tetroxide, OsO4. They complement one another in that the first is used in aqueous solution whereas the latter is used in organic solvents. The mechanisms are very similar. Thus both proceed through a pericyclic mechanism: O O O O Mn(VII) Mn(V) +- K+-O O K O O a manganate ester hydrolysis O OH HO Mn(IV)O2 + KOH + 0.5 H2O2 Mn(V) + +- K O OH HO O O O O Os(VIII) Os(VI) O O O O an osmate ester, yellow black Because osmium tetroxide is expensive and very toxic it is rarely used in stoichiometric amounts. Rather a co-oxidant is added to oxidise the stable osmate ester back to OsO4. Since we want to hydrolyse the osmate ester as well, the co-oxidant is often aqueous hydrogen peroxide so the oxidation is two-phase. The alkene and the OsO4 remain in the organic phase, the H2O2 is in the aqueous phase and the osmate ester is at the interface between the two phases: H OH H OH H OsO4, + aq.H2O2, CH Cl H 2 2 OH H H OH (c) Epoxidation (X = Y = O) The reaction of alkenes with peroxy acids (RCO3H) leads to a cyclic ether, known as an epoxide, as follows: R R O O O H O H O O Although the reaction will work with peroxyacetic acid (R = Me) it works best with peroxy acids bearing electron-withdrawing groups e.g. -

Potassium Permanganate MSDS
He a lt h 2 0 Fire 0 1 0 Re a c t iv it y 0 Pe rs o n a l Pro t e c t io n J Material Safety Data Sheet Potassium permanganate MSDS Section 1: Chemical Product and Company Identification Product Name: Potassium permanganate Contact Information: Catalog Codes: SLP4912, SLP3892, SLP1075 Sciencelab.com, Inc. 14025 Smith Rd. CAS#: 7722-64-7 Houston, Texas 77396 RTECS: SD6475000 US Sales: 1-800-901-7247 International Sales: 1-281-441-4400 TSCA: TSCA 8(b) inventory: Potassium permanganate Order Online: ScienceLab.com CI#: Not available. CHEMTREC (24HR Emergency Telephone), call: Synonym: Potassium Permanganate, Biotech Grade 1-800-424-9300 Chemical Name: Potassium Permanganate International CHEMTREC, call: 1-703-527-3887 Chemical Formula: KMnO4 For non-emergency assistance, call: 1-281-441-4400 Section 2: Composition and Information on Ingredients Composition: Name CAS # % by Weight Potassium permanganate 7722-64-7 100 Toxicological Data on Ingredients: Potassium permanganate, Biotech: ORAL (LD50): Acute: 1090 mg/kg [Rat]. 2157 mg/kg [Mouse]. Section 3: Hazards Identification Potential Acute Health Effects: Hazardous in case of skin contact (irritant), of eye contact (irritant), of ingestion, of inhalation. Slightly hazardous in case of skin contact (permeator). Possibly corrosive to eyes and skin. The amount of tissue damage depends on length of contact. Eye contact can result in corneal damage or blindness. Skin contact can produce inflammation and blistering. Inhalation of dust will produce irritation to gastro-intestinal or respiratory tract, characterized by burning, sneezing and coughing. Severe over-exposure can produce lung damage, choking, unconsciousness or death. -

In Situ Chemical Oxidation of Creosote/Coal Tar Residuals: Experimental and Numerical Investigation
In situ Chemical Oxidation of Creosote/Coal Tar Residuals: Experimental and Numerical Investigation by Steven Philip Forsey A thesis presented to the University of Waterloo in fulfillment of the thesis requirement of the degree of Doctor of Philosophy in Earth Sciences Waterloo, Ontario, Canada, 2004 ©Steven Philip Forsey 2004 I herby declare that I am the sole author of this thesis. This is a true copy of the thesis, including any required final version, as accepted by my examiners. I understand that my thesis may be made electronically available to the public Steven Forsey ii Abstract Coal tar, coal tar creosote and oily wastes are often present as subsurface contaminants that may migrate below the water table, leaving a widely distributed residual source of contaminants leaching to the ground water. In situ chemical oxidation is a potentially viable technology for the remediation of aquifers contaminated with creosote and coal tars. The oxidant of choice would be flushed through the contaminated area to oxidize aqueous contaminants and enhance the mass transfer of contaminants from the oil phase. A series of batch and column experiments were performed to assess the ability of a chemical oxidizing reagent to oxidize creosote compounds and to increase mass transfer rates. Results from the column experiments were then simulated using a reactive transport model that considered 12 different creosote compounds undergoing dissolution, oxidation and advective-dispersive transport. Three strong chemical oxidizing reagents, Fenton’s Reagent, potassium persulfate with ferrous ions, and potassium permanganate were tested with batch experiments to determine their reactivity towards creosote compounds. All three reagents successfully decomposed aqueous creosote compounds and were able to reduce the mass of the monitored creosote compounds within the oil phase. -

The Anodic Oxidation of Maleic Acid
Scholars' Mine Masters Theses Student Theses and Dissertations 1966 The anodic oxidation of maleic acid Larry D. Gilmartin Follow this and additional works at: https://scholarsmine.mst.edu/masters_theses Part of the Chemical Engineering Commons Department: Recommended Citation Gilmartin, Larry D., "The anodic oxidation of maleic acid" (1966). Masters Theses. 5776. https://scholarsmine.mst.edu/masters_theses/5776 This thesis is brought to you by Scholars' Mine, a service of the Missouri S&T Library and Learning Resources. This work is protected by U. S. Copyright Law. Unauthorized use including reproduction for redistribution requires the permission of the copyright holder. For more information, please contact [email protected]. THE ANODIC OXIDATION OF MALEIC ACID BY LARRY D. GILMARTIN A THESIS submitted to the faculty of THE UNIVERSITY OF MISSOURI AT ROLLA in partial fulfillment of the requirements for the Degree of MASTER OF SCIENCE IN CHEMICAL ENGINEERING Rolla, Missouri 1966 Approved by (advisor) THE ANODIC OXIDATION OF MALEIC ACID Larry D. Gilmartin ABSTRACT The purpose of this investigation was to determine the mechanism of the anodic oxidation of maleic acid on platinized-platinum electrodes at 80°C. Current density-potential studies were conducted varying the parameters of maleic acid concentration and pH. The faradaic efficiency of the oxidation of maleic acid to yield co 2 was determined. The effect of temperature on current density was also studied to determine the activation energy for the reaction. The oxidation of maleic acid occurred only in acidic solutions. The faradaic efficiency was found to be ap proximately 97 ± 5 per cent. A linear Tafel region was found which had a slope of 145 - 170 millivolts <~ 2.3RT/aF). -

Complexes of Ferrous Iron with Tannic Acid Fy J
Complexes of Ferrous Iron With Tannic Acid fy J. D. HEM :HEMISTRY OF IRON IN NATURAL WATER GEOLOGICAL SURVEY WATER-SUPPLY PAPER 1459-D IITED STATES GOVERNMENT PRINTING OFFICE, WASHINGTON : 1960 UNITED STATES DEPARTMENT OF THE INTERIOR FRED A. SEATON, Secretary GEOLOGICAL SURVEY Thomas B. Nolan, Director For sale by the Superintendent of Documents, U.S. Government Printing Office Washington 25, D.C. CONTENTS Page Abstract. _________________________________________________________ 75 Acknowledgments. ________________________________________________ 75 Organic complexing agents________-______-__-__-__-______-____-___-- 75 Tannic acid_______________________________________________________ 77 Properties ____________________________________________________ 78 Dissociation._________________________________________________ 78 Reducing action_____--_-______________________________________ 79 Laboratory studies_______________________________________________ 79 Analytical procedures__________________________________________ 80 Chemical reactions in test solutions._____________________________ 81 No tannic acid____________________-_________________-_--__ 84 Five parts per million of tannic acid- ________________________ 84 Fifty parts per million of tannic acid_____-________-____------ 85 Five hundred parts per million of tanni c acid _________________ 86 Rate of oxidation and precipitation of iron______________________ 87 Stability constants for tannic acid complexes______________________ 88 Comparison of determined and estimated Eh______________________ -

Chemical Incompatibilities
Chemical Incompatibilities Incompatibilities Chromic acid, nitric acid, hydroxyl compounds, ethylene glycol, Acetic acid perchloric acid, peroxides, permanganates Acetylene Chlorine, bromine, copper, fluorine, silver, mercury Acetone Concentrated nitric and sulfuric acid mixtures Alkali and alkaline earth metals (such as Water, carbon tetrachloride or other chlorinated hydrocarbons, powdered aluminum or magnesium, calcium, carbon dioxide, halogens lithium, sodium, potassium) Mercury (in manometers, for example), chlorine, calcium Ammonia (anhydrous) hypochlorite, iodine, bromine, hydrofluoric acid (anhydrous) Acids, powdered metals, flammable liquids, chlorates, nitrites, Ammonium nitrate sulfur, finely divided organic combustible materials Aniline Nitric acid, hydrogen peroxide Arsenical materials Any reducing agent Azides Acids Bromine See chlorine Calcium oxide Water Carbon (activated) Calcium hypochlorite, all oxidizing agents Carbon tetrachloride Sodium Ammonium salts, acids, powdered metals, sulfur, finely divided Chlorates organic or combustible materials Acetic acid, naphthalene, camphor, glycerol, alcohol, flammable Chromic acid and chromium liquids in general Ammonia, acetylene, butadiene, butane, methane, propane (or Chlorine other petroleum gases), hydrogen, sodium carbide, benzene, finely divided metals, turpentine Chlorine dioxide Ammonia, methane, phosphine, hydrogen sulfide Copper Acetylene, hydrogen peroxide Cumene hydroperoxide Acids (organic or inorganic) Cyanides Acids Ammonium nitrate, chromic acid, hydrogen peroxide,