Xagrid, INN-Anagrelide
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Photoaging & Skin Damage
Use_for_Revised_OFC_Only_2006_PhotoagingSkinDamage 5/21/13 9:11 AM Page 2 PEORIA (309) 674-7546 MORTON (309) 263-7546 GALESBURG (309) 344-5777 PERU (815) 224-7400 NORMAL (309) 268-9980 CLINTON, IA (563) 242-3571 DAVENPORT, IA (563) 344-7546 SoderstromSkinInstitute.comsoderstromskininstitute.com FROMFrom YOUR Your DERMATOLOGISTDermatologist [email protected]@skinnews.com PHOTOAGING & SKIN DAMAGE Before You Worship The Sun Who’s At Risk? Today, many researchers and dermatologists Skin types that burn easily and tan rarely are believe that wrinkling and aging changes of the skin much more susceptible to the ravages of the sun on the are much more related to sun damage than to age! skin than are those that tan easily, rather than burn. Many of the signs of skin damage from the sun are Light complected, blue-eyed, red-haired people such as pictured on these pages. The decrease in the ozone Swedish, Irish, and English, are usually more suscep- layer, increasing the sun’s intensity, and the increasing tible to photo damage, and their skin shows the signs sun exposure among our population – through work, of photo damage earlier in life and in a more pro- sports, sunbathing and tanning parlors – have taken a nounced manner. Dark complexions give more protec- tremendous toll on our skin. Sun damage to the skin tion from light and the sun. ranks with other serious health dangers of smoking, alcohol, and increased cholesterol, and is being seen in younger and younger people. NO TAN IS A SAFE TAN! Table of Contents Sun Damage .............................................Pg. 1 Skin Cancer..........................................Pgs. 2-3 Mohs Micrographic Surgery ......................Pg. -

Late Stent Thrombosis After Paclitaxel-Eluting Stent Placement in a Patient with Essential Thrombocytosis
558 Türk Kardiyol Dern Arş - Arch Turk Soc Cardiol 2010;38(8):558-560 Late stent thrombosis after paclitaxel-eluting stent placement in a patient with essential thrombocytosis Esansiyel trombositozlu bir hastada paklitaksel salınımlı stent yerleştirme sonrası gelişen geç stent trombozu Telat Keleş, M.D., Nihal Akar Bayram, M.D., Tahir Durmaz, M.D., Engin Bozkurt, M.D. Department of Cardiology, Ankara Atatürk Education and Research Hospital, Ankara We report on a case of late stent thrombosis after drug- Bu yazıda, esansiyel trombositozlu bir hastada ilaç sa- eluting stent placement in a patient with essential throm- lınımlı stent yerleştirme sonrası gelişen geç stent trom- bocytosis. A 51-year-old male patient with a three-month bozu sunuldu. Üç ay önce sol ön inen artere paklitak- history of paclitaxel-eluting stent placement to the left sel salınımlı stent yerleştirilen 51 yaşındaki erkek hasta anterior descending artery presented with a complaint şiddetli retrosternal göğüs ağrısı yakınmasıyla başvur- of severe retrosternal chest pain. A high platelet count du. İki ay öncesinde hastanın trombosit sayımı yüksek (1,063,000/mm3) was detected two months prior to (1063000/mm3) bulunmuş ve durumu esansiyel trombo- presentation, which was interpreted as essential throm- sitoz olarak yorumlanmıştı. Hasta standart ikili antitrom- bocytosis. He was on standard dual antiplatelet therapy bosit tedavi (aspirin ve klopidogrel) görmekteydi. Elekt- (aspirin and clopidogrel). The electrocardiogram showed rokardiyografide V1-V6 derivasyonlarında ST-segment ST-segment elevation in leads V1-V6. Emergent coro- yükselmesi izlendi. Acil koroner anjiyografide paklitaksel nary angiography revealed thrombotic total occlusion salınımlı stent yerinde trombotik tam tıkanıklık gözlendi. at the location of the paclitaxel-eluting stent. -

May 2020 for Health Professionals Who Care for Cancer Patients
Systemic Therapy Update Volume 23 Issue 5 May 2020 For Health Professionals Who Care for Cancer Patients Inside This Issue: Editor’s Choice Benefit Drug List New Programs: Neoadjuvant Carboplatin for Triple-Negative New: BRLACTWAC, UGUPAPA, Pegaspargase (LKNOS, Breast Cancer (BRLACTWAC) | Apalutamide for Non- LYNOS), Bevacizumab (Pediatric), IGEV (Pediatric) | Metastatic Castration-Resistant Prostate Cancer (UGUPAPA) Deleted: GIGAVEOCAP, GIGAVEOF, SMAJIFN Drug Update NEW Protocols, PPPOs and Patient Handouts Pharmacare Update – Anticoagulants | Patient Assistance BR: BRLACTWAC | GU: UGUPAPA Programs REVISED Protocols, PPPOs and Patient Handouts Drug Shortages BR: BRAJAC, BRAJACT, BRAJACTG, BRAJACTT, BRAJACTTG, New: Alemtuzumab, Anagrelide | Updated: Hydroxyurea BRAJACTW, BRAJDAC, BRAJFEC, BRAJFECD, BRAJFECDT, Cancer Drug Manual© BRAVAC, BRLAACD, BRLAACDT, BRLATACG, BRLATWAC | New: Abemaciclib, Lurbinectedin | Revised: Apalutamide, GI: GIPGEMABR | GU: UGUPABI, UGUPENZ | LK: ULKBLIN, Durvalumab, Panitumumab, Pembrolizumab ULKMDSA | SA: SAIME | SC: SCDRUGRX | SM: USMAVDAB, ST Update Editorial Board USMAVDT, USMAVTRA, USMAVVC, USMAVVEM Membership Update Resources and Contact Information Editor’s Choice New Programs Effective 01 May 2020, the BC Cancer Provincial Systemic Therapy Program has approved the following new treatment programs. The full details of these programs can be found on the BC Cancer website in the Chemotherapy Protocols section. Breast Neoadjuvant Carboplatin for Locally Advanced, Triple-Negative Breast Cancer (BRLACTWAC) — The Provincial Systemic Therapy Program has approved the addition of carboplatin to the standard taxane- anthracycline neoadjuvant chemotherapy regimens, for patients with stage II and higher triple-negative breast cancer. This regimen consists of carboplatin (given every 3 weeks) in combination with weekly paclitaxel, followed by doxorubicin-cyclophosphamide (AC given every 2 or 3 weeks). Filgrastim support is available for the dose-dense AC option. -

BC Cancer Benefit Drug List September 2021
Page 1 of 65 BC Cancer Benefit Drug List September 2021 DEFINITIONS Class I Reimbursed for active cancer or approved treatment or approved indication only. Reimbursed for approved indications only. Completion of the BC Cancer Compassionate Access Program Application (formerly Undesignated Indication Form) is necessary to Restricted Funding (R) provide the appropriate clinical information for each patient. NOTES 1. BC Cancer will reimburse, to the Communities Oncology Network hospital pharmacy, the actual acquisition cost of a Benefit Drug, up to the maximum price as determined by BC Cancer, based on the current brand and contract price. Please contact the OSCAR Hotline at 1-888-355-0355 if more information is required. 2. Not Otherwise Specified (NOS) code only applicable to Class I drugs where indicated. 3. Intrahepatic use of chemotherapy drugs is not reimbursable unless specified. 4. For queries regarding other indications not specified, please contact the BC Cancer Compassionate Access Program Office at 604.877.6000 x 6277 or [email protected] DOSAGE TUMOUR PROTOCOL DRUG APPROVED INDICATIONS CLASS NOTES FORM SITE CODES Therapy for Metastatic Castration-Sensitive Prostate Cancer using abiraterone tablet Genitourinary UGUMCSPABI* R Abiraterone and Prednisone Palliative Therapy for Metastatic Castration Resistant Prostate Cancer abiraterone tablet Genitourinary UGUPABI R Using Abiraterone and prednisone acitretin capsule Lymphoma reversal of early dysplastic and neoplastic stem changes LYNOS I first-line treatment of epidermal -

Phosphodiesterase (PDE)
Phosphodiesterase (PDE) Phosphodiesterase (PDE) is any enzyme that breaks a phosphodiester bond. Usually, people speaking of phosphodiesterase are referring to cyclic nucleotide phosphodiesterases, which have great clinical significance and are described below. However, there are many other families of phosphodiesterases, including phospholipases C and D, autotaxin, sphingomyelin phosphodiesterase, DNases, RNases, and restriction endonucleases, as well as numerous less-well-characterized small-molecule phosphodiesterases. The cyclic nucleotide phosphodiesterases comprise a group of enzymes that degrade the phosphodiester bond in the second messenger molecules cAMP and cGMP. They regulate the localization, duration, and amplitude of cyclic nucleotide signaling within subcellular domains. PDEs are therefore important regulators ofsignal transduction mediated by these second messenger molecules. www.MedChemExpress.com 1 Phosphodiesterase (PDE) Inhibitors, Activators & Modulators (+)-Medioresinol Di-O-β-D-glucopyranoside (R)-(-)-Rolipram Cat. No.: HY-N8209 ((R)-Rolipram; (-)-Rolipram) Cat. No.: HY-16900A (+)-Medioresinol Di-O-β-D-glucopyranoside is a (R)-(-)-Rolipram is the R-enantiomer of Rolipram. lignan glucoside with strong inhibitory activity Rolipram is a selective inhibitor of of 3', 5'-cyclic monophosphate (cyclic AMP) phosphodiesterases PDE4 with IC50 of 3 nM, 130 nM phosphodiesterase. and 240 nM for PDE4A, PDE4B, and PDE4D, respectively. Purity: >98% Purity: 99.91% Clinical Data: No Development Reported Clinical Data: No Development Reported Size: 1 mg, 5 mg Size: 10 mM × 1 mL, 10 mg, 50 mg (R)-DNMDP (S)-(+)-Rolipram Cat. No.: HY-122751 ((+)-Rolipram; (S)-Rolipram) Cat. No.: HY-B0392 (R)-DNMDP is a potent and selective cancer cell (S)-(+)-Rolipram ((+)-Rolipram) is a cyclic cytotoxic agent. (R)-DNMDP, the R-form of DNMDP, AMP(cAMP)-specific phosphodiesterase (PDE) binds PDE3A directly. -

ER Guide to Bleeding Disorders
Bleeding disorders ER guide to bleeding disorders 1 Table of contents 4 General Guidelines 4–5 national Hemophilia Foundation guidelines 5–10 Treatment options 10 HemopHilia a Name:__________________________________________________________________________________________________ 10–11 national Hemophilia Foundation guidelines Address:________________________________________________________________________________________________ 12 dosage chart Phone:__________________________________________________________________________________________________ 14–15 Treatment products 16 HemopHilia B In case of emergency, contact: ______________________________________________________________________________ 16 national Hemophilia Foundation guidelines Relation to patient:________________________________________________________________________________________ 17 dosage chart 18 Treatment products 19 HemopHilia a or B with inHiBiTors Diagnosis: Hemophilia A: Mild Moderate Severe 20 national Hemophilia Foundation guidelines Inhibitors Inhibitors Bethesda units (if known) ____________________________________ 21 Treatment products Hemophilia B: Mild Moderate Severe 22–23 Von willeBrand disease Inhibitors Inhibitors Bethesda units (if known) ____________________________________ 23–24 national Hemophilia Foundation guidelines von Willebrand disease: Type 1 Type 2 Type 3 Platelet type 25 Treatment products 27 Bibliography Preferred product:_________________________________________________________________________________________ Dose for life-threatening -
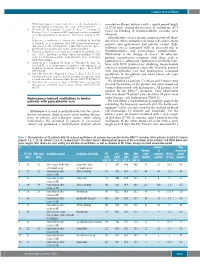
Hydroxyurea Induced Oscillations in Twelve Patients with Polycythemia
Letters to the Editor JAK2 haplotype is a major risk factor for the development of revealed oscillatory behavior with a typical period length myeloproliferative neoplasms. Nat Genet. 2009;41(4):446-9. of 27-30 days. During the presence of oscillations (25.5 6. Olcaydu D, Harutyunyan A, Jager R, Berg T, Gisslinger B, Pabinger I, et al. A common JAK2 haplotype confers susceptibil- years) no bleeding or thromboembolic episodes were ity to myeloproliferative neoplasms. Nat Genet. 2009;41(4):450- observed. 4. Polycythemia vera is a chronic acquired stem cell disor- 7. Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally der which affects primarily red blood cells and in many A, Ebert BL, et al. A germline JAK2 SNP is associated with pre- patients also granulocyte and platelet counts. Poly - disposition to the development of JAK2V617F-positive myelo- cythemia vera is associated with an increased risk of proliferative neoplasms. Nat Genet. 2009;41(4):455-9. 1 8. Olcaydu D, Skoda RC, Looser R, Li S, Cazzola M, Pietra D, et al. thromboembolic and hemorrhagic complications. The 'GGCC' haplotype of JAK2 confers susceptibility to JAK2 Phlebotomy is the therapy of choice.2 In refractory exon 12 mutation-positive polycythemia vera. Leukemia. patients cytoreductive treatment with drugs such as 2009;23(10):1924-6. hydroxyurea is advocated. Hydroxyurea primarily inter- 9. Harrison CN, Campbell PJ, Buck G, Wheatley K, East CL, Bareford D, et al. Hydroxyurea Compared with Anagrelide in feres with DNA synthesis by inhibiting ribonucleotide 3 High-Risk Essential Thrombocythemia. N Engl J Med. 2005; reductase in hematopoietic stem cells. -

Pharmaceuticals As Environmental Contaminants
PharmaceuticalsPharmaceuticals asas EnvironmentalEnvironmental Contaminants:Contaminants: anan OverviewOverview ofof thethe ScienceScience Christian G. Daughton, Ph.D. Chief, Environmental Chemistry Branch Environmental Sciences Division National Exposure Research Laboratory Office of Research and Development Environmental Protection Agency Las Vegas, Nevada 89119 [email protected] Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada Why and how do drugs contaminate the environment? What might it all mean? How do we prevent it? Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada This talk presents only a cursory overview of some of the many science issues surrounding the topic of pharmaceuticals as environmental contaminants Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada A Clarification We sometimes loosely (but incorrectly) refer to drugs, medicines, medications, or pharmaceuticals as being the substances that contaminant the environment. The actual environmental contaminants, however, are the active pharmaceutical ingredients – APIs. These terms are all often used interchangeably Office of Research and Development National Exposure Research Laboratory, Environmental Sciences Division, Las Vegas, Nevada Office of Research and Development Available: http://www.epa.gov/nerlesd1/chemistry/pharma/image/drawing.pdfNational -

(12) Patent Application Publication (10) Pub. No.: US 2009/0246185 A1 Kishida Et Al
US 20090246185A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2009/0246185 A1 Kishida et al. (43) Pub. Date: Oct. 1, 2009 (54) CARDIAC DYSFUNCTION-AMELIORATING (30) Foreign Application Priority Data AGENT OR CARDAC FUNCTION-MANTAININGAGENT Mar. 13, 2006 (JP) ................................. 2006-066992 Nov. 21, 2006 (JP) ................................. 2006-314034 (75) Inventors: Hideyuki Kishida, Hyogo (JP); O O Kenji Fujii, Hyogo (JP); Hiroshi Publication Classification Kubo, Hyogo (JP); Kazunori (51) Int. Cl. Hosoe, Hyogo (JP) A6II 3L/22 (2006.01) CD7C 43/23 (2006.01) Correspondence Address: A6IP 9/00 (2006.01) SUGHRUE MION, PLLC (52) U.S. Cl. ........................................ 424/94.1:568/651 2100 PENNSYLVANIA AVENUE, N.W., SUITE 8OO (57) ABSTRACT WASHINGTON, DC 20037 (US) An object of the present invention is to provide a highly safe oral composition Superior in a cardiac dysfunction-amelio (73) Assignee: KANEKA CORPORATION, rating or cardiac function-maintaining action. The present OSAKA-SHI, OSAKA (JP) inventors have conducted intensive studies in an attempt to solve the aforementioned problems and found that use of (21) Appl. No.: 12/282,448 particularly, reduced coenzyme Q10 from among highly safe coenzyme Q affords a composition useful for amelioration of (22) PCT Filed: Mar. 9, 2007 cardiac dysfunction and maintenance of cardiac function. Accordingly, the present invention provides a cardiac dys (86). PCT No.: PCT/UP2007/O54.643 function-ameliorating agent or cardiac function-maintaining agent containing reduced coenzyme Qas an active ingredient, S371 (c)(1), and a pharmaceutical product, a food, an animal drug, a feed (2), (4) Date: Dec. 23, 2008 and the like, which contain the agent. -

Customs Tariff - Schedule
CUSTOMS TARIFF - SCHEDULE 99 - i Chapter 99 SPECIAL CLASSIFICATION PROVISIONS - COMMERCIAL Notes. 1. The provisions of this Chapter are not subject to the rule of specificity in General Interpretative Rule 3 (a). 2. Goods which may be classified under the provisions of Chapter 99, if also eligible for classification under the provisions of Chapter 98, shall be classified in Chapter 98. 3. Goods may be classified under a tariff item in this Chapter and be entitled to the Most-Favoured-Nation Tariff or a preferential tariff rate of customs duty under this Chapter that applies to those goods according to the tariff treatment applicable to their country of origin only after classification under a tariff item in Chapters 1 to 97 has been determined and the conditions of any Chapter 99 provision and any applicable regulations or orders in relation thereto have been met. 4. The words and expressions used in this Chapter have the same meaning as in Chapters 1 to 97. Issued January 1, 2019 99 - 1 CUSTOMS TARIFF - SCHEDULE Tariff Unit of MFN Applicable SS Description of Goods Item Meas. Tariff Preferential Tariffs 9901.00.00 Articles and materials for use in the manufacture or repair of the Free CCCT, LDCT, GPT, UST, following to be employed in commercial fishing or the commercial MT, MUST, CIAT, CT, harvesting of marine plants: CRT, IT, NT, SLT, PT, COLT, JT, PAT, HNT, Artificial bait; KRT, CEUT, UAT, CPTPT: Free Carapace measures; Cordage, fishing lines (including marlines), rope and twine, of a circumference not exceeding 38 mm; Devices for keeping nets open; Fish hooks; Fishing nets and netting; Jiggers; Line floats; Lobster traps; Lures; Marker buoys of any material excluding wood; Net floats; Scallop drag nets; Spat collectors and collector holders; Swivels. -

Preoperative Medication Directive a Clinical Practice Protocol Practitioner Information
Preoperative Medication Directive A Clinical Practice Protocol Practitioner Information Please note: this is a living document subject to change. We recommend that this not be printed but rather accessed on line for the most current information. Nov 2017 Table of Contents Regional practice protocol for Preoperative Medication Page Directives Purpose & Intent 1 Practice Outcomes 1 Background 2 Components 2 References 3 Authors 4 Guidelines- Section A (for select Nurses) 5 Guidelines- Section B (for Surgeons) 12 Guidelines- Section C (for Anaesthesiologists) pending PURPOSE AND INTENT Who this document is for: These sectioned documents are a resource and an information source for Preoperative Assessment/Anaesthesia Clinic Staff, Surgeons and Anaesthesiologists. Each Section (A, B, etc...) was created for a primary user group. Users of this document shall always employ critical thinking and seek additional guidance from the appropriate colleagues (ie: anaesthesiologist and/or surgeon/prescribing physician) when concerns or variances arise. 1. Practice Outcomes How to use this document: This tool is to supplement the practice at each site. Implementation of this document will be at the discretion of the site leaders, however the directions provided to patients regarding preoperative medications should be consistent with this document (under the consideration of critical thinking). Clinical judgment, critical thinking and patient specific needs & concerns should always be employed, considered and addressed for all parts of the document. Any care provider with a Preoperative Medication Directive medication question is asked to submit in writing the medication in question (along with any supporting evidence if available) to the PAC Working Group (chair) [email protected] for consideration. -

Nivedita Singh
Xanthine Based Inhibitors for Therapeutics Targeting Phosphodiesterase 9A A Thesis Submitted in Partial Fulfilment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY BY NIVEDITA SINGH DEPARTMENT OF BIOSCIENCES & BIOENGINEERING INDIAN INSTITUTE OF TECHNOLOGY GUWAHATI GUWAHATI-781039, ASSAM, INDIA NOVEMBER 2016 Xanthine Based Inhibitors for Therapeutics Targeting Phosphodiesterase 9A A Thesis Submitted in Partial Fulfilment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY BY NIVEDITA SINGH DEPARTMENT OF BIOSCIENCES & BIOENGINEERING INDIAN INSTITUTE OF TECHNOLOGY GUWAHATI GUWAHATI-781039, ASSAM, INDIA NOVEMBER 2016 TH-1894_11610615 TH-1894_11610615 INDIAN INSTITUTE OF TECHNOLOGY GUWAHATI DEPARTMENT OF BIOSCIENCE & BIOENGINEERING STATEMENT I do hereby declare that the matter embodied in this thesis entitled: “Xanthine based inhibitors for therapeutics targeting phosphodiesterase 9A”, is the result of investigations carried out by me in the Department of Biosciences & Bioengineering, Indian Institute of Technology Guwahati, India, under the guidance of Dr. Sanjukta Patra and co-supervision of Prof. Parameswar Krishnan Iyer. In keeping with the general practice of reporting scientific observations, due acknowledgements have been made wherever the work described is based on the findings of other investigators. November, 2016 Ms. Nivedita Singh Roll No. 11610615 TH-1894_11610615 INDIAN INSTITUTE OF TECHNOLOGY GUWAHATI DEPARTMENT OF BIOSCIENCE & BIOENGINEERING CERTIFICATE It is certified that the work described in this thesis, entitled: “Xanthine based inhibitors for therapeutics targeting phosphodiesterase 9A”, done by Ms Nivedita Singh (Roll No: 11610615), for the award of degree of Doctor of Philosophy is an authentic record of the results obtained from the research work carried out under my supervision in the Department of Biosciences & Bioengineering, Indian Institute of Technology Guwahati, India, and this work has not been submitted elsewhere for a degree.