More Than 18000 Effectors in the Legionella Genus Genome Provide Multiple, Independent Combinations for Replication In
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Characterization of Aminoacyl-Trna Synthetases in Chromerids
Article Characterization of Aminoacyl-tRNA Synthetases in Chromerids Abdoallah Sharaf 1,2, Ansgar Gruber 1, Kateřina Jiroutová 1 and Miroslav Oborník 1,3,* 1 Institute of Parasitology, Biology Centre, Czech Academy of Sciences, 370 05 České Budějovice, Czech Republic 2 Genetics Department, Faculty of Agriculture, Ain Shams University, Cairo 11241, Egypt 3 Faculty of Science, University of South Bohemia, 370 05 České Budějovice, Czech Republic * Correspondence: [email protected] Received: 1 July 2019; Accepted: 28 July 2019; Published: 31 July 2019 Abstract: Aminoacyl-tRNA synthetases (AaRSs) are enzymes that catalyze the ligation of tRNAs to amino acids. There are AaRSs specific for each amino acid in the cell. Each cellular compartment in which translation takes place (the cytosol, mitochondria, and plastids in most cases), needs the full set of AaRSs; however, individual AaRSs can function in multiple compartments due to dual (or even multiple) targeting of nuclear- encoded proteins to various destinations in the cell. We searched the genomes of the chromerids, Chromera velia and Vitrella brassicaformis, for AaRS genes: 48 genes encoding AaRSs were identified in C. velia, while only 39 AaRS genes were found in V. brassicaformis. In the latter alga, ArgRS and GluRS were each encoded by a single gene occurring in a single copy; only PheRS was found in three genes, while the remaining AaRSs were encoded by two genes. In contrast, there were nine cases for which C. velia contained three genes of a given AaRS (45% of the AaRSs), all of them representing duplicated genes, except AsnRS and PheRS, which are more likely pseudoparalogs (acquired via horizontal or endosymbiotic gene transfer). -

Oborník M.& Lukeš, J. (2013) Cell Biology of Chromerids: Autotrophic
CHAPTER EIGHT Cell Biology of Chromerids: Autotrophic Relatives to Apicomplexan Parasites Miroslav Oborník*,†,{,1, Julius Lukeš*,† *Biology Centre, Institute of Parasitology, Academy of Sciences of the Czech Republic, Cˇ eske´ Budeˇjovice, Czech Republic †Faculty of Science, University of South Bohemia, Cˇ eske´ Budeˇjovice, Czech Republic { Institute of Microbiology, Academy of Sciences of the Czech Republic, Trˇebonˇ, Czech Republic 1Corresponding author: e-mail address: [email protected] Contents 1. Introduction 334 2. Chromerida: A New Group of Algae Isolated from Australian Corals 337 2.1 C. velia: A new alga from Sydney Harbor 338 2.2 V. brassicaformis: An alga from the Great Barrier Reef 343 3. Life Cycle 346 4. Evolution of Exosymbiont 348 5. Evolution of Chromerid Organelles 350 5.1 Evolution of chromerid plastids 350 5.2 Reduced mitochondrial genomes of chromerids 354 5.3 Chromerosome: C. velia as a possible mixotroph 354 6. Metabolism of Chromerids 355 6.1 Unique pathway for tetrapyrrole biosynthesis 355 6.2 Other metabolic features of C. velia 359 7. Chromerids as Possible Symbionts of Corals 361 8. Conclusions 361 Acknowledgments 362 References 362 Abstract Chromerida are algae possessing a complex plastid surrounded by four membranes. Although isolated originally from stony corals in Australia, they seem to be globally dis- tributed. According to their molecular phylogeny, morphology, ultrastructure, structure of organellar genomes, and noncanonical pathway for tetrapyrrole synthesis, these algae are thought to be the closest known phototrophic relatives to apicomplexan par- asites. Here, we summarize the current knowledge of cell biology and evolution of this novel group of algae, which contains only two formally described species, but is appar- ently highly diverse and virtually ubiquitous in marine environments. -

(PAD) and Post-Translational Protein Deimination—Novel Insights Into Alveolata Metabolism, Epigenetic Regulation and Host–Pathogen Interactions
biology Article Peptidylarginine Deiminase (PAD) and Post-Translational Protein Deimination—Novel Insights into Alveolata Metabolism, Epigenetic Regulation and Host–Pathogen Interactions Árni Kristmundsson 1,*, Ásthildur Erlingsdóttir 1 and Sigrun Lange 2,* 1 Institute for Experimental Pathology at Keldur, University of Iceland, Keldnavegur 3, 112 Reykjavik, Iceland; [email protected] 2 Tissue Architecture and Regeneration Research Group, School of Life Sciences, University of Westminster, London W1W 6UW, UK * Correspondence: [email protected] (Á.K.); [email protected] (S.L.) Simple Summary: Alveolates are a major group of free living and parasitic organisms; some of which are serious pathogens of animals and humans. Apicomplexans and chromerids are two phyla belonging to the alveolates. Apicomplexans are obligate intracellular parasites; that cannot complete their life cycle without exploiting a suitable host. Chromerids are mostly photoautotrophs as they can obtain energy from sunlight; and are considered ancestors of the apicomplexans. The pathogenicity and life cycle strategies differ significantly between parasitic alveolates; with some causing major losses in host populations while others seem harmless to the host. As the life cycles of Citation: Kristmundsson, Á.; Erlingsdóttir, Á.; Lange, S. some are still poorly understood, a better understanding of the factors which can affect the parasitic Peptidylarginine Deiminase (PAD) alveolates’ life cycles and survival is of great importance and may aid in new biomarker discovery. and Post-Translational Protein This study assessed new mechanisms relating to changes in protein structure and function (so-called Deimination—Novel Insights into “deimination” or “citrullination”) in two key parasites—an apicomplexan and a chromerid—to Alveolata Metabolism, Epigenetic assess the pathways affected by this protein modification. -

The Organellar Genomes of Chromera and Vitrella, the Phototrophic
MI69CH07-Lukes ARI 5 June 2015 13:47 V I E E W R S I E N C N A D V A The Organellar Genomes of Chromera and Vitrella,the Phototrophic Relatives of Apicomplexan Parasites Miroslav Obornık´ 1,2,3 and Julius Lukesˇ1,2,4 1Institute of Parasitology, Biology Center, Czech Academy of Sciences, 1160/31 Ceskˇ e´ Budejovice,ˇ Czech Republic; email: [email protected], [email protected] 2Faculty of Science, University of South Bohemia, 37005 Ceskˇ e´ Budejovice,ˇ Czech Republic 3Institute of Microbiology, Czech Academy of Sciences, 379 81 Treboˇ n,ˇ Czech Republic 4Canadian Institute for Advanced Research, Toronto, Ontario M5G 1Z8, Canada Annu. Rev. Microbiol. 2015. 69:129–44 Keywords The Annual Review of Microbiology is online at organellar genomes, mitochondrion, plastid, Apicomplexa, Alveolata, micro.annualreviews.org Chromera This article’s doi: 10.1146/annurev-micro-091014-104449 Abstract Copyright c 2015 by Annual Reviews. Apicomplexa are known to contain greatly reduced organellar genomes. All rights reserved Their mitochondrial genome carries only three protein-coding genes, and their plastid genome is reduced to a 35-kb-long circle. The discovery of coral- endosymbiotic algae Chromera velia and Vitrella brassicaformis, which share a common ancestry with Apicomplexa, provided an opportunity to study possibly ancestral forms of organellar genomes, a unique glimpse into the evolutionary history of apicomplexan parasites. The structurally similar mi- tochondrial genomes of Chromera and Vitrella differ in gene content, which is reflected in the composition of their respiratory chains. Thus, Chromera lacks respiratory complexes I and III, whereas Vitrella and apicomplexan parasites are missing only complex I. -

Light Harvesting Complexes of Chromera Velia, Photosynthetic Relative of Apicomplexan Parasites
Biochimica et Biophysica Acta 1827 (2013) 723–729 Contents lists available at SciVerse ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbabio Light harvesting complexes of Chromera velia, photosynthetic relative of apicomplexan parasites Josef Tichy a,b, Zdenko Gardian a,b, David Bina a,b, Peter Konik a, Radek Litvin a,b, Miroslava Herbstova a,b, Arnab Pain c, Frantisek Vacha a,b,⁎ a Faculty of Science, University of South Bohemia, Branisovska 31, 37005 Ceske Budejovice, Czech Republic b Institute of Plant Molecular Biology, Biology Centre ASCR, Branisovska 31, 37005 Ceske Budejovice, Czech Republic c Computational Bioscience Research Center, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia article info abstract Article history: The structure and composition of the light harvesting complexes from the unicellular alga Chromera velia Received 19 October 2012 were studied by means of optical spectroscopy, biochemical and electron microscopy methods. Two different Received in revised form 31 January 2013 types of antennae systems were identified. One exhibited a molecular weight (18–19 kDa) similar to FCP Accepted 5 February 2013 (fucoxanthin chlorophyll protein) complexes from diatoms, however, single particle analysis and circular Available online 18 February 2013 dichroism spectroscopy indicated similarity of this structure to the recently characterized XLH antenna of “ ” Keywords: xanthophytes. In light of these data we denote this antenna complex CLH, for Chromera Light Harvesting fi Chromera velia complex. The other system was identi ed as the photosystem I with bound Light Harvesting Complexes FCP (PSI–LHCr) related to the red algae LHCI antennae. The result of this study is the finding that C. -
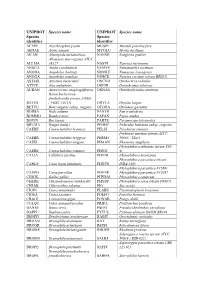
Additional File 1 Revised.Pdf
UNIPROT Species name UNIPROT Species name Species Species identifier identifier ACYPI Acyrthosiphon pisum MUSPF Mustela putorius furo AEDAE Aedes aegypti MYOLU Myotis lucifugus AILME Ailuropoda melanoleuca NAEGR Naegleria gruberi Allomyces macrogynus ATCC ALLMA 38327 NASVI Nasonia vitripennis ANOCA Anolis carolinensis NEMVE Nematostella vectensis ANODA Anopheles darlingi NOMLE Nomascus leucogenys ANOGA Anopheles gambiae NOSCE Nosema ceranae (strain BRL01) ASTMX Astyanax mexicanus ONCVO Onchocerca volvulus ATTCE Atta cephalotes ORENI Oreochromis niloticus AURAN Aureococcus anophagefferens ORNAN Ornithorhynchus anatinus Batrachochytrium dendrobatidis (strain JAM81 BATDJ / FGSC 10211) ORYLA Oryzias latipes BETVU Beta vulgaris subsp. vulgaris OTOGA Otolemur garnettii BODSA Bodo saltans PANTR Pan troglodytes BOMMO Bombyx mori PAPAN Papio anubis BOVIN Bos taurus PARTE Paramecium tetraurelia BRUMA Brugia malayi PEDHC Pediculus humanus subsp. corporis CAEBE Caenorhabditis brenneri PELSI Pelodiscus sinensis Perkinsus marinus (strain ATCC CAEBR Caenorhabditis briggsae PERM5 50983 / TXsc) CAEEL Caenorhabditis elegans PHAAN Phaseolus angularis Phytophthora infestans (strain T30- CAERE Caenorhabditis remanei PHYIT 4) CALJA Callithrix jacchus PHYNI Phytophthora nicotianae Phytophthora parasitica (strain CANLF Canis lupus familiaris PHYPN INRA-310) Phytophthora parasitica P1569/ CAVPO Cavia porcellus PHYPR Phytophthora parasitica P10297 CHICK Gallus gallus PHYRM Phytophthora ramorum CHLRE Chlamydomonas reinhardtii PHYSP Phytophthora sojae (strain P6497) -

Phylogenetics of Cladopyxidoiddinophytes
www.nature.com/scientificreports OPEN Fensomea setacea, gen. & sp. nov. (Cladopyxidaceae, Dinophyceae), is neither gonyaulacoid nor peridinioid as inferred from morphological and molecular data Marc Gottschling1, Maria Consuelo Carbonell‑Moore2, Kenneth Neil Mertens3, Monika Kirsch4, Malte Elbrächter5 & Urban Tillmann6* Dinophyte evolution is essentially inferred from the pattern of thecal plates, and two diferent labelling systems are used for the important subgroups Gonyaulacales and Peridiniales. The partiform hypotheca of cladopyxidoid dinophytes fts into the morphological concepts of neither group, although they are assigned to the Gonyaulacales. Here, we describe the thecate dinophyte Fensomea setacea, gen. & sp. nov., which has a cladopyxidoid tabulation. The cells displayed a Kofoidean plate formula APC, 3′, 4a, 7″, 7C, 6S, 6′′′, 2′′′′, and slender processes were randomly distributed over the echinate or baculate surface. In addition, we obtained rRNA sequences of F. setacea, gen. & sp. nov., but dinophytes that exhibit a partiform hypotheca did not show a close relationship to Gonyaulacales. Character evolution of thecate dinophytes may have progressed from the ancestral state of six postcingular plates, and two more or less symmetrically arranged antapical plates, towards patterns of only fve postcingular plates (Peridiniales) or more asymmetrical confgurations (Gonyaulacales). Based on our phylogenetic reconsiderations the contact between the posterior sulcal plate and the frst postcingular plate, as well as the contact between an antapical plate and the distalmost postcingular plate, do not represent a rare, specialized gonyaulacoid plate confguration (i.e., the partiform hypotheca of cladopyxidoid dinophytes). Instead, these contacts correspond to the common and regular confguration of peridinioid (and other) dinophytes. Over time, evolution has produced impressive biodiversity in the world’s oceans, including multicellular organ- isms, such as animals, and unicellular organisms that make signifcant ecological contributions 1,2. -

A Method for Identification of Highly Conserved Elements and Evolutionary Analysis of Superphylum Alveolata Lev I
Rubanov et al. BMC Bioinformatics (2016) 17:385 DOI 10.1186/s12859-016-1257-5 RESEARCH ARTICLE Open Access A method for identification of highly conserved elements and evolutionary analysis of superphylum Alveolata Lev I. Rubanov*, Alexandr V. Seliverstov, Oleg A. Zverkov and Vassily A. Lyubetsky Abstract Background: Perfectly or highly conserved DNA elements were found in vertebrates, invertebrates, and plants by various methods. However, little is known about such elements in protists. The evolutionary distance between apicomplexans can be very high, in particular, due to the positive selection pressure on them. This complicates the identification of highly conserved elements in alveolates, which is overcome by the proposed algorithm. Results: A novel algorithm is developed to identify highly conserved DNA elements. It is based on the identification of dense subgraphs in a specially built multipartite graph (whose parts correspond to genomes). Specifically, the algorithm does not rely on genome alignments, nor pre-identified perfectly conserved elements; instead, it performs a fast search for pairs of words (in different genomes) of maximum length with the difference below the specified edit distance. Such pair defines an edge whose weight equals the maximum (or total) length of words assigned to its ends. The graph composed of these edges is then compacted by merging some of its edges and vertices. The dense subgraphs are identified by a cellular automaton-like algorithm; each subgraph defines a cluster composed of similar inextensible words from different genomes. Almost all clusters are considered as predicted highly conserved elements. The algorithm is applied to the nuclear genomes of the superphylum Alveolata, and the corresponding phylogenetic tree is built and discussed. -

A Comparative Overview of the Flagellar Apparatus of Dinoflagellate, Perkinsids and Colpodellids
Microorganisms 2014, 2, 73-91; doi:10.3390/microorganisms2010073 OPEN ACCESS microorganisms ISSN 2076-2607 www.mdpi.com/journal/microorganisms Review A Comparative Overview of the Flagellar Apparatus of Dinoflagellate, Perkinsids and Colpodellids Noriko Okamoto and Patrick J. Keeling * Centre for Microbial Diversity and Evolution, Department of Botany, University of British Columbia, 3529-6270 University Boulevard, Vancouver, BC V6T 1Z4, Canada; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-604-822-2845; Fax: +1-604-822-6089. Received: 24 November 2013; in revised form: 29 January 2014 / Accepted: 8 February 2014 / Published: 10 March 2014 Abstract: Dinoflagellates are a member of the Alveolata, and elucidation of the early evolution of alveolates is important for our understanding of dinoflagellates, and vice versa. The ultrastructure of the flagellar apparatus has been described from several dinoflagellates in the last few decades, and the basic components appear to be well conserved. The typical dinoflagellate apparatus is composed of two basal bodies surrounded by striated collars attached to a connective fiber. The longitudinal basal body is connected to a longitudinal microtubular root (LMR; equivalent of R1) and single microtubular root (R2), whereas the transverse basal body is connected to a transverse microtubular root (TMR; R3) and transverse striated root (TSR) with a microtubule (R4). Some of these components, especially the connective fibers and collars, are dinoflagellate specific characteristics that make their flagellar apparatus relatively complex. We also compare these structures with the flagellar apparatus from a number of close relatives of dinoflagellates and their sister, the apicomplexans, including colpodellids, perkinsids, and Psammosa. -

Dinoflagellate Plastids Waller and Koreny Revised
Running title: Plastid complexity in dinoflagellates Title: Plastid complexity in dinoflagellates: a picture of gains, losses, replacements and revisions Authors: Ross F Waller and Luděk Kořený Affiliations: Department of Biochemistry, University of Cambridge, Cambridge, CB2 1QW, UK Keywords: endosymbiosis, plastid, reductive evolution, mixotrophy, kleptoplast, Myzozoa, peridinin Abstract Dinoflagellates are exemplars of plastid complexity and evolutionary possibility. Their ordinary plastids are extraordinary, and their extraordinary plastids provide a window into the processes of plastid gain and integration. No other plastid-bearing eukaryotic group possesses so much diversity or deviance from the basic traits of this cyanobacteria-derived endosymbiont. Although dinoflagellate plastids provide a major contribution to global carbon fixation and energy cycles, they show a remarkable willingness to tinker, modify and dispense with canonical function. The archetype dinoflagellate plastid, the peridinin plastid, has lost photosynthesis many times, has the most divergent organelle genomes of any plastid, is bounded by an atypical plastid membrane number, and uses unusual protein trafficking routes. Moreover, dinoflagellates have gained new endosymbionts many times, representing multiple different stages of the processes of organelle formation. New insights into dinoflagellate plastid biology and diversity also suggests it is timely to revise notions of the origin of the peridinin plastid. I. Introduction Dinoflagellates represent a major plastid-bearing protist lineage that diverged from a common ancestor shared with apicomplexan parasites (Figure 1). Since this separation dinoflagellates have come to exploit a wide range of marine and aquatic niches, providing critical environmental services at a global level, as well as having significant negative impact on some habitats and communities. As photosynthetic organisms, dinoflagellates contribute a substantial fraction of global carbon fixation that drives food webs as well as capture of some anthropogenic CO2. -

Fatty Acid Biosynthesis in Chromerids
biomolecules Article Fatty Acid Biosynthesis in Chromerids 1,2, 1,3, 1,3 1 Aleš Tomˇcala y , Jan Michálek y, Ivana Schneedorferová , Zoltán Füssy , Ansgar Gruber 1 , Marie Vancová 1 and Miroslav Oborník 1,3,* 1 Biology Centre CAS, Institute of Parasitology, Branišovská 31, 370 05 Ceskˇ é Budˇejovice,Czech Republic; [email protected] (A.T.); [email protected] (J.M.); [email protected] (I.S.); [email protected] (Z.F.); [email protected] (A.G.); [email protected] (M.V.) 2 Faculty of Fisheries and Protection of Waters, CENAKVA, Institute of Aquaculture and Protection of Waters, University of South Bohemia, Husova 458/102, 370 05 Ceskˇ é Budˇejovice,Czech Republic 3 Faculty of Science, University of South Bohemia, Branišovská 31, 370 05 Ceskˇ é Budˇejovice,Czech Republic * Correspondence: [email protected]; Tel.: +420-38777-5464 These authors equally contributed to this work. y Received: 14 May 2020; Accepted: 15 July 2020; Published: 24 July 2020 Abstract: Fatty acids are essential components of biological membranes, important for the maintenance of cellular structures, especially in organisms with complex life cycles like protozoan parasites. Apicomplexans are obligate parasites responsible for various deadly diseases of humans and livestock. We analyzed the fatty acids produced by the closest phototrophic relatives of parasitic apicomplexans, the chromerids Chromera velia and Vitrella brassicaformis, and investigated the genes coding for enzymes involved in fatty acids biosynthesis in chromerids, in comparison to their parasitic relatives. Based on evidence from genomic and metabolomic data, we propose a model of fatty acid synthesis in chromerids: the plastid-localized FAS-II pathway is responsible for the de novo synthesis of fatty acids reaching the maximum length of 18 carbon units. -

CAFE Estimations of Expansions and Contractions in the Apicomplexan
Supplementary Figures and Tables Supplementary Figure 1: CAFE estimations of expansions and contractions in the apicomplexan sugar transporter gene family The phylogenetic tree shows the mean number of sugar transporters gained or lost per node. Minus indicates a net contraction. Nodes are numbered in angle brackets. The phylogenetic tree was generated by a Bayesian approach using representative sugar transporter protein sequences from clades 1 and 2 (Figure 2). Supplementary Table 1: Species information and sequence sources Species Strain NCBI Sequence Database URL Database Taxon ID Source Version Babesia bigemina BOND 5866 PiroplasmaDB http://piroplasmadb.org release 25 Babesia bovis T2Bo 484906 PiroplasmaDB http://piroplasmadb.org release 25 Babesia microti RI 1133968 PiroplasmaDB http://piroplasmadb.org release 25 Theileria annulata Ankara 353154 PiroplasmaDB http://piroplasmadb.org release 25 Theileria equi WA 1537102 PiroplasmaDB http://piroplasmadb.org release 25 Theileria orientalis Shintoku 869250 PiroplasmaDB http://piroplasmadb.org release 25 18 Theileria parva Muguga 333668 PiroplasmaDB http://piroplasmadb.org release 25 Cryptosporidium TU502 353151 CryptoDB http://cryptodb.org release 25 hominis Cryptosporidium muris RN66 441375 CryptoDB http://cryptodb.org release 25 Cryptosporidium Iowa II 353152 CryptoDB http://cryptodb.org release 25 parvum Gregarina niphandrodes unknown 110365 CryptoDB http://cryptodb.org release 25 Chromera velia CCMP2878 1169474 CryptoDB http://cryptodb.org release 25 Vitrella brassicaformis CCMP3155