Structureenergybased Predictions and Network Modelling of Rasopathy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DGCR8 Microprocessor Defect Characterizes Familial Multinodular Goiter with Schwannomatosis
The Journal of Clinical Investigation CLINICAL MEDICINE DGCR8 microprocessor defect characterizes familial multinodular goiter with schwannomatosis Barbara Rivera,1,2,3 Javad Nadaf,2,3 Somayyeh Fahiminiya,4 Maria Apellaniz-Ruiz,2,3,4,5 Avi Saskin,5,6 Anne-Sophie Chong,2,3 Sahil Sharma,7 Rabea Wagener,8 Timothée Revil,5,9 Vincenzo Condello,10 Zineb Harra,2,3 Nancy Hamel,4 Nelly Sabbaghian,2,3 Karl Muchantef,11,12 Christian Thomas,13 Leanne de Kock,2,3,5 Marie-Noëlle Hébert-Blouin,14 Angelia V. Bassenden,15 Hannah Rabenstein,8 Ozgur Mete,16,17 Ralf Paschke,18,19,20,21,22 Marc P. Pusztaszeri,23 Werner Paulus,13 Albert Berghuis,15 Jiannis Ragoussis,4,9 Yuri E. Nikiforov,10 Reiner Siebert,8 Steffen Albrecht,24 Robert Turcotte,25,26 Martin Hasselblatt,13 Marc R. Fabian,1,2,3,7,15 and William D. Foulkes1,2,3,4,5,6 1Gerald Bronfman Department of Oncology, McGill University, Montreal, Quebec, Canada. 2Lady Davis Institute for Medical Research and 3Segal Cancer Centre, Jewish General Hospital, Montreal, Quebec, Canada. 4Cancer Research Program, McGill University Health Centre, Montreal, Quebec, Canada. 5Department of Human Genetics, McGill University, Montreal, Quebec, Canada. 6Division of Medical Genetics, Department of Medicine, McGill University Health Centre and Jewish General Hospital, Montreal, Quebec, Canada. 7Department of Experimental Medicine, McGill University, Montreal, Quebec, Canada. 8Institute of Human Genetics, University of Ulm and University of Ulm Medical Center, Ulm, Germany. 9Génome Québec Innovation Centre, McGill University, Montreal, Quebec, Canada. 10Department of Pathology, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, USA. 11Department of Diagnostic Radiology, McGill University, Montreal, Quebec, Canada. -

Identification of Transcriptional Mechanisms Downstream of Nf1 Gene Defeciency in Malignant Peripheral Nerve Sheath Tumors Daochun Sun Wayne State University
Wayne State University DigitalCommons@WayneState Wayne State University Dissertations 1-1-2012 Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors Daochun Sun Wayne State University, Follow this and additional works at: http://digitalcommons.wayne.edu/oa_dissertations Recommended Citation Sun, Daochun, "Identification of transcriptional mechanisms downstream of nf1 gene defeciency in malignant peripheral nerve sheath tumors" (2012). Wayne State University Dissertations. Paper 558. This Open Access Dissertation is brought to you for free and open access by DigitalCommons@WayneState. It has been accepted for inclusion in Wayne State University Dissertations by an authorized administrator of DigitalCommons@WayneState. IDENTIFICATION OF TRANSCRIPTIONAL MECHANISMS DOWNSTREAM OF NF1 GENE DEFECIENCY IN MALIGNANT PERIPHERAL NERVE SHEATH TUMORS by DAOCHUN SUN DISSERTATION Submitted to the Graduate School of Wayne State University, Detroit, Michigan in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY 2012 MAJOR: MOLECULAR BIOLOGY AND GENETICS Approved by: _______________________________________ Advisor Date _______________________________________ _______________________________________ _______________________________________ © COPYRIGHT BY DAOCHUN SUN 2012 All Rights Reserved DEDICATION This work is dedicated to my parents and my wife Ze Zheng for their continuous support and understanding during the years of my education. I could not achieve my goal without them. ii ACKNOWLEDGMENTS I would like to express tremendous appreciation to my mentor, Dr. Michael Tainsky. His guidance and encouragement throughout this project made this dissertation come true. I would also like to thank my committee members, Dr. Raymond Mattingly and Dr. John Reiners Jr. for their sustained attention to this project during the monthly NF1 group meetings and committee meetings, Dr. -

S41467-021-24841-Y.Pdf
ARTICLE https://doi.org/10.1038/s41467-021-24841-y OPEN Integrative oncogene-dependency mapping identifies RIT1 vulnerabilities and synergies in lung cancer Athea Vichas1,10, Amanda K. Riley 1,2,10, Naomi T. Nkinsi1, Shriya Kamlapurkar1, Phoebe C. R. Parrish 1,3, April Lo1,3, Fujiko Duke4, Jennifer Chen4, Iris Fung4, Jacqueline Watson4, Matthew Rees 4, Austin M. Gabel 3,5,6,7, James D. Thomas 6,7, Robert K. Bradley 3,6,7, John K. Lee1, Emily M. Hatch 1,7, ✉ Marina K. Baine8, Natasha Rekhtman8, Marc Ladanyi8, Federica Piccioni4,9 & Alice H. Berger 1,3 1234567890():,; CRISPR-based cancer dependency maps are accelerating advances in cancer precision medicine, but adequate functional maps are limited to the most common oncogenes. To identify opportunities for therapeutic intervention in other rarer subsets of cancer, we investigate the oncogene-specific dependencies conferred by the lung cancer oncogene, RIT1. Here, genome-wide CRISPR screening in KRAS, EGFR, and RIT1-mutant isogenic lung cancer cells identifies shared and unique vulnerabilities of each oncogene. Combining this genetic data with small-molecule sensitivity profiling, we identify a unique vulnerability of RIT1- mutant cells to loss of spindle assembly checkpoint regulators. Oncogenic RIT1M90I weakens the spindle assembly checkpoint and perturbs mitotic timing, resulting in sensitivity to Aurora A inhibition. In addition, we observe synergy between mutant RIT1 and activation of YAP1 in multiple models and frequent nuclear overexpression of YAP1 in human primary RIT1-mutant lung tumors. These results provide a genome-wide atlas of oncogenic RIT1 functional inter- actions and identify components of the RAS pathway, spindle assembly checkpoint, and Hippo/YAP1 network as candidate therapeutic targets in RIT1-mutant lung cancer. -

Genetic Testing in Inherited Endocrine Disorders
Eggermann et al. Orphanet Journal of Rare Diseases (2020) 15:144 https://doi.org/10.1186/s13023-020-01420-w REVIEW Open Access Genetic testing in inherited endocrine disorders: joint position paper of the European reference network on rare endocrine conditions (Endo-ERN) Thomas Eggermann1* , Miriam Elbracht1, Ingo Kurth1, Anders Juul2,3, Trine Holm Johannsen2,3, Irène Netchine4, George Mastorakos5, Gudmundur Johannsson6, Thomas J. Musholt7, Martin Zenker8, Dirk Prawitt9, Alberto M. Pereira10, Olaf Hiort11 and on behalf of the European Reference Network on Rare Endocrine Conditions (ENDO-ERN Abstract Background: With the development of molecular high-throughput assays (i.e. next generation sequencing), the knowledge on the contribution of genetic and epigenetic alterations to the etiology of inherited endocrine disorders has massively expanded. However, the rapid implementation of these new molecular tools in the diagnostic settings makes the interpretation of diagnostic data increasingly complex. Main body: This joint paper of the ENDO-ERN members aims to overview chances, challenges, limitations and relevance of comprehensive genetic diagnostic testing in rare endocrine conditions in order to achieve an early molecular diagnosis. This early diagnosis of a genetically based endocrine disorder contributes to a precise management and helps the patients and their families in their self-determined planning of life. Furthermore, the identification of a causative (epi)genetic alteration allows an accurate prognosis of recurrence risks for family planning as the basis of genetic counselling. Asymptomatic carriers of pathogenic variants can be identified, and prenatal testing might be offered, where appropriate. Conclusions: The decision on genetic testing in the diagnostic workup of endocrine disorders should be based on their appropriateness to reliably detect the disease-causing and –modifying mutation, their informational value, and cost-effectiveness. -

2016 Nf Conference
2016 NF CONFERENCE “YOU’VE GOT THE POWER!” June 18-21, 2016 JW Marriott Austin, TX NF CONFERENCE 2 | Children’s Tumor Foundation · Ending Neurofibromatosis Through Research Dear NF Conference Attendees: Welcome to Austin—the capital of Texas, the live music capital of the world, and for these next three and a half days, the capital of NF research! We are thrilled to be with you all in such a thriving city, an ideal location for the greatest minds in NF research to congregate, collaborate, and make strides towards ending NF. This year’s 2016 NF Conference, brief neighbors of the NF Patient Forum, is based on the larger premise that you, along with the incredibly brave NF patients and their families, have the power to end NF. There are three core principles of the NF Conference that drive this premise: creating new networks and friendships for attendees; bringing patients, researchers and clinicians face- to-face, and ensuring that we at the Foundation continue to use this event as a platform to stimulate collaborative NF research, and to showcase our incredible progress. Moreover, at this Conference, there will be the first open data release in NF. It is only as a team that we can succeed! It is you, our friends and colleagues, and your dedication to NF research, who have made this progress and promise a reality. You represent the expansive, and often unpredictable spectrum that encompasses science, and within the singularly difficult realm of NF research. It is you who have made all of the accomplishments in NF since last year’s Conference possible, and it is you who will foster further accomplishments this year and beyond. -

The Ezrin Metastatic Phenotype Is Associated with the Initiation of Protein Translation
Volume 14 Number 4 April 2012 pp. 297–310 297 www.neoplasia.com The Ezrin Metastatic Phenotype Is Joseph W. Briggs*, Ling Ren*, Rachel Nguyen*, Kristi Chakrabarti*, Jessica Cassavaugh*, Associated with the Initiation of Said Rahim†, Gulay Bulut†, Ming Zhou‡, 1 Timothy D. Veenstra‡, Qingrong Chen§, Jun S. Wei§, Protein Translation Javed Khan§, Aykut Uren† and Chand Khanna* *Tumor and Metastasis Biology Section, Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA; †Lombardi Cancer Center, Georgetown University, Washington, DC, USA; ‡Laboratory of Proteomics and Analytical Technologies, Advanced Technology Program, SAIC – Frederick, Inc, National Cancer Institute - Frederick, Frederick, MD, USA; §Oncogenomics Section, Pediatric Oncology Branch, Center for Cancer Research, National Cancer Institute, Bethesda, MD, USA Abstract We previously associated the cytoskeleton linker protein, Ezrin, with the metastatic phenotype of pediatric sarcomas, including osteosarcoma and rhabdomyosarcoma. These studies have suggested that Ezrin contributes to the survival of cancer cells after their arrival at secondary metastatic locations. To better understand this role in metastasis, we undertook two noncandidate analyses of Ezrin function including a microarray subtraction of high- and low-Ezrin- expressing cells and a proteomic approach to identify proteins that bound the N-terminus of Ezrin in tumor lysates. Functional analyses of these data led to a novel and unifying hypothesis that Ezrin contributes to the efficiency of metastasis through regulation of protein translation. In support of this hypothesis, we found Ezrin to be part of the ribonucleoprotein complex to facilitate the expression of complex messenger RNA in cells and to bind with poly A binding protein 1 (PABP1; PABPC1). -

Genetic and Epigenetic Mechanisms in the Development of Congenital Heart Diseases
Open access Review World Jnl Ped Surgery: first published as 10.1136/wjps-2020-000196 on 29 April 2021. Downloaded from Genetic and epigenetic mechanisms in the development of congenital heart diseases Yue Wu,1 Xiaosi Jin,2 Yuhao Zhang,3 Jing Zheng,1 Rulai Yang1 To cite: Wu Y, Jin X, Zhang Y, ABSTRACT surgical intervention techniques and peri- et al. Genetic and epigenetic Congenital heart disease (CHD) is the most common operative care has dramatically changed the mechanisms in the development of congenital cardiovascular malformations associated management of these populations with CHD. of congenital heart diseases. with birth defects, and it results in significant morbidity World Jnl Ped Surgery However, CHD is still a bothersome question and mortality worldwide. The classification of CHD is 2021;4:e000196. doi:10.1136/ owing to its undesirable outcomes and expen- still elusive owing to the complex pathogenesis of CHD. wjps-2020-000196 sive healthcare costs, which bring substantial Advances in molecular medicine have revealed the genetic physiological, emotional and socioeconomic ► Additional supplemental basis of some heart anomalies. Genes associated with material is published online only. CHD might be modulated by various epigenetic factors. challenges to patients, families and society. To view, please visit the journal Thus, the genetic and epigenetic factors are gradually According to the final anatomical and online (http:// dx. doi. org/ 10. 1136/ accepted as important triggers in the pathogenesis of pathophysiological complexities, CHD can wjps- 2020- 000196). CHD. However, few literatures have comprehensively be classified as mild, moderate or severe. 4 elaborated the genetic and epigenetic mechanisms of CHD. -

MAFB Determines Human Macrophage Anti-Inflammatory
MAFB Determines Human Macrophage Anti-Inflammatory Polarization: Relevance for the Pathogenic Mechanisms Operating in Multicentric Carpotarsal Osteolysis This information is current as of October 4, 2021. Víctor D. Cuevas, Laura Anta, Rafael Samaniego, Emmanuel Orta-Zavalza, Juan Vladimir de la Rosa, Geneviève Baujat, Ángeles Domínguez-Soto, Paloma Sánchez-Mateos, María M. Escribese, Antonio Castrillo, Valérie Cormier-Daire, Miguel A. Vega and Ángel L. Corbí Downloaded from J Immunol 2017; 198:2070-2081; Prepublished online 16 January 2017; doi: 10.4049/jimmunol.1601667 http://www.jimmunol.org/content/198/5/2070 http://www.jimmunol.org/ Supplementary http://www.jimmunol.org/content/suppl/2017/01/15/jimmunol.160166 Material 7.DCSupplemental References This article cites 69 articles, 22 of which you can access for free at: http://www.jimmunol.org/content/198/5/2070.full#ref-list-1 by guest on October 4, 2021 Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2017 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. -

LZTR1 Inactivation Promotes MAPK/ ERK Pathway Activation
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.14.989954; this version posted March 15, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. LZTR1 inactivation promotes MAPK/ ERK pathway activation in glioblastoma by stabilizing oncoprotein RIT1 Running Title: LZTR1 regulates RIT1 stability Keywords: glioblastoma; gene mutations; LZTR1; RIT1; ubiquitination; degradation; MAPK/ERK pathway Yuqi Wang1, #, Jianong Zhang2, #, Pingzhao Zhang3, #, Zhipeng Zhao1, #, Qilin Huang4, Dapeng Yun5, Juxiang Chen4, Hongyan Chen1, Chenji Wang1, *, Daru Lu1,6, * 1State Key Laboratory of Genetic Engineering and MOE Engineering Research Center of Gene Technology, School of Life Sciences, Fudan University, Shanghai, 200438, China, 2Obstetrics & Gynecology Hospital of Fudan University, State Key Lab of Genetic Engineering, School of Life Sciences and Institutes of Biomedical Sciences, Shanghai 200438, China, 3Fudan University Shanghai Cancer Center and Institutes of Biomedical Sciences, Shanghai Medical College, Fudan University, Shanghai 200032, China, 4Department of Neurosurgery, Shanghai Institute of Neurosurgery, Changzheng Hospital, Second Military Medical University, Shanghai, 200003, China, 5Department of Pharmacology, The University of Texas Southwestern Medical Center, Dallas, TX75390, USA, 6Key Laboratory of Birth Defects and Reproductive Health of National Health and Family Planning CommissionChongqing Population and Family PlanningScience and Technology Research Institute, Chongqing 400020China # These authors contributed equally to this work. *Corresponding Author: Daru Lu, E-mail: [email protected]; Chenji Wang, E-mail: [email protected]. ABSTRACT Large-scale sequencing studies on glioblastoma have identified numerous genetic alterations. Leucine-zipper-like transcription regulator 1 (LZTR1) is inactivated by non-synonymous mutations and copy number losses, suggesting that it is a tumor suppressor in glioblastoma. -

Gene Expression Mechanism and Repertoire of ASC-Mediated
The Journal of Immunology Mechanism and Repertoire of ASC-Mediated Gene Expression1 Mizuho Hasegawa,2* Ryu Imamura,* Kou Motani,* Takumi Nishiuchi,† Norihiko Matsumoto,* Takeshi Kinoshita,* and Takashi Suda3* Apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) is an adaptor molecule that mediates inflammatory and apoptotic signals. Although the role of ASC in caspase-1-mediated IL-1 and IL-18 maturation is well known, ASC also induces NF-B activation and cytokine gene expression in human cells. In this study, we investigated the molecular mechanism and repertoire of ASC-induced gene expression in human cells. We found that the specific activation of ASC induced AP-1 activity, which was required for optimal IL8 promoter activity. ASC activation also induced STAT3-, but not STAT1-, IFN-stimulated gene factor 3- or NF-AT-dependent reporter gene expression. The ASC-mediated AP-1 activation was NF-B-independent and primarily cell-autonomous response, whereas the STAT3 activation required NF-B activation and was mediated by a factor that can act in a paracrine manner. ASC-mediated AP-1 activation was inhibited by chemical or protein inhibitors for caspase-8, caspase-8-targeting small-interfering RNA, and p38 and JNK inhibitors, but not by a caspase-1 inhibitor, caspase-9 or Fas-associated death domain protein (FADD) dominant-negative mutants, FADD- or RICK-targeting small-interfering RNAs, or a MEK inhibitor, indicating that the ASC-induced AP-1 activation is mediated by caspase-8, p38, and JNK, but does not require caspase-1, caspase-9, FADD, RICK, or ERK. DNA microarray analyses identified 75 genes that were induced by ASC activation. -
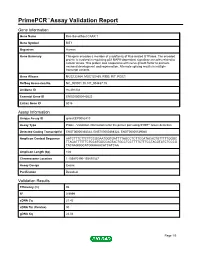
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name Ras-like without CAAX 1 Gene Symbol RIT1 Organism Human Gene Summary This gene encodes a member of a subfamily of Ras-related GTPases. The encoded protein is involved in regulating p38 MAPK-dependent signaling cascades related to cellular stress. This protein also cooperates with nerve growth factor to promote neuronal development and regeneration. Alternate splicing results in multiple transcript variants. Gene Aliases MGC125864, MGC125865, RIBB, RIT, ROC1 RefSeq Accession No. NC_000001.10, NT_004487.19 UniGene ID Hs.491234 Ensembl Gene ID ENSG00000143622 Entrez Gene ID 6016 Assay Information Unique Assay ID qHsaCEP0052410 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000368323, ENST00000368322, ENST00000539040 Amplicon Context Sequence AATCTTTCTTCTTCCGGAATGGTGATTTTAGCCTCTTCCATACACTGTTTTTGGGC TTAGATTTTTTCTCCATGGCCAGTACTGCCTCCTTTTCTTTCCTACGTATCTCCCG TACAAGGGCATGGAAAACATCATCAA Amplicon Length (bp) 108 Chromosome Location 1:155870190-155870327 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 96 R2 0.9999 cDNA Cq 21.43 cDNA Tm (Celsius) 80 gDNA Cq 24.04 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report RIT1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of -

Bcl11b Controls Odorant Receptor Class Choice in Mice
Corrected: Author Correction ARTICLE https://doi.org/10.1038/s42003-019-0536-x OPEN Bcl11b controls odorant receptor class choice in mice Takayuki Enomoto1,2, Hidefumi Nishida2, Tetsuo Iwata1, Akito Fujita2, Kanako Nakayama2, Takahiro Kashiwagi2, Yasue Hatanaka1, Hiro Kondo2, Rei Kajitani 2, Takehiko Itoh 2, Makoto Ohmoto1,3, Ichiro Matsumoto3 & Junji Hirota 1,2 1234567890():,; Each olfactory sensory neuron (OSN) expresses a single odorant receptor (OR) gene from the class I or class II repertoire in mice. The mechanisms that regulate OR class choice in OSNs remain unknown. Here, we show that the transcription factor Bcl11b determines the OR class to be expressed in OSNs. Both loss- and gain-of-function analyses demonstrate that class I is a default fate of OSNs and that Bcl11b dictates a class II OR choice by suppressing the effect of the J-element, a class I-OR enhancer. We further demonstrate that OSN-specific genetic manipulations of Bcl11b bias the OR class choice, generating mice with “class I- dominant” and “class II-dominant” noses, which display contrasting innate olfactory beha- viors to two distinct aversive odorants. Overall, these findings reveal a unique transcriptional mechanism mediating a binary switch for OR class choice that is crucial to both the ana- tomical and functional organization of the olfactory system. 1 Center for Biological Resources and Informatics, Tokyo Institute of Technology, Yokohama 226-8501, Japan. 2 Department of Life Science and Technology, Graduate School of Life Science and Technology, Tokyo Institute of Technology, Yokohama 226-8501, Japan. 3 Monell Chemical Senses Center, Philadelphia, PA 19104, USA. Correspondence and requests for materials should be addressed to J.H.