ARGLU1 Is an RNA-Binding Protein That Has a Fundamental Role in Modulating Global Alternative Splicing Patterns
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Association Study for Circulating Fibroblast Growth Factor
www.nature.com/scientificreports OPEN Genome‑wide association study for circulating fbroblast growth factor 21 and 23 Gwo‑Tsann Chuang1,2,14, Pi‑Hua Liu3,4,14, Tsui‑Wei Chyan5, Chen‑Hao Huang5, Yu‑Yao Huang4,6, Chia‑Hung Lin4,7, Jou‑Wei Lin8, Chih‑Neng Hsu8, Ru‑Yi Tsai8, Meng‑Lun Hsieh9, Hsiao‑Lin Lee9, Wei‑shun Yang2,9, Cassianne Robinson‑Cohen10, Chia‑Ni Hsiung11, Chen‑Yang Shen12,13 & Yi‑Cheng Chang2,9,12* Fibroblast growth factors (FGFs) 21 and 23 are recently identifed hormones regulating metabolism of glucose, lipid, phosphate and vitamin D. Here we conducted a genome‑wide association study (GWAS) for circulating FGF21 and FGF23 concentrations to identify their genetic determinants. We enrolled 5,000 participants from Taiwan Biobank for this GWAS. After excluding participants with diabetes mellitus and quality control, association of single nucleotide polymorphisms (SNPs) with log‑transformed FGF21 and FGF23 serum concentrations adjusted for age, sex and principal components of ancestry were analyzed. A second model additionally adjusted for body mass index (BMI) and a third model additionally adjusted for BMI and estimated glomerular fltration rate (eGFR) were used. A total of 4,201 participants underwent GWAS analysis. rs67327215, located within RGS6 (a gene involved in fatty acid synthesis), and two other SNPs (rs12565114 and rs9520257, located between PHC2-ZSCAN20 and ARGLU1-FAM155A respectively) showed suggestive associations with serum FGF21 level (P = 6.66 × 10–7, 6.00 × 10–7 and 6.11 × 10–7 respectively). The SNPs rs17111495 and rs17843626 were signifcantly associated with FGF23 level, with the former near PCSK9 gene and the latter near HLA-DQA1 gene (P = 1.04 × 10–10 and 1.80 × 10–8 respectively). -

Controls Homeostatic Splicing of ARGLU1 Mrna Stephan P
Published online 28 November 2016 Nucleic Acids Research, 2017, Vol. 45, No. 6 3473–3486 doi: 10.1093/nar/gkw1140 An Ultraconserved Element (UCE) controls homeostatic splicing of ARGLU1 mRNA Stephan P. Pirnie, Ahmad Osman, Yinzhou Zhu and Gordon G. Carmichael* Department of Genetics and Genome Sciences, UCONN Health Center, 400 Farmington Avenue, Farmington, CT 06030, USA Received January 13, 2016; Revised October 25, 2016; Editorial Decision October 28, 2016; Accepted October 31, 2016 ABSTRACT or inhibit the usage of a particular splice site (1,2). The ex- pression of trans-acting factors in a developmental and tis- Arginine and Glutamate-Rich protein 1 (ARGLU1) is sue specific manner results in regulated splicing that isof- a protein whose function is poorly understood, but ten cell specific. Most notably, trans-acting proteins such as may act in both transcription and pre-mRNA splicing. the NOVA (3–6), the RBFOX (7) and SR protein (8–11) We demonstrate that the ARGLU1 gene expresses families, and a number of hnRNP (12,13) proteins compete at least three distinct RNA splice isoforms – a fully to bind nascent RNAs at specific motifs and drive regula- spliced isoform coding for the protein, an isoform tion of alternative splicing in a tissue and developmentally containing a retained intron that is detained in the specific manner. Alternative splicing is an important pro- nucleus, and an isoform containing an alternative cess that is seen in at least 95% of multi-exon genes in the exon that targets the transcript for nonsense medi- human transcriptome (14). Furthermore, alternative splic- ated decay. -

Reconstructing Cell Cycle Pseudo Time-Series Via Single-Cell Transcriptome Data—Supplement
School of Natural Sciences and Mathematics Reconstructing Cell Cycle Pseudo Time-Series Via Single-Cell Transcriptome Data—Supplement UT Dallas Author(s): Michael Q. Zhang Rights: CC BY 4.0 (Attribution) ©2017 The Authors Citation: Liu, Zehua, Huazhe Lou, Kaikun Xie, Hao Wang, et al. 2017. "Reconstructing cell cycle pseudo time-series via single-cell transcriptome data." Nature Communications 8, doi:10.1038/s41467-017-00039-z This document is being made freely available by the Eugene McDermott Library of the University of Texas at Dallas with permission of the copyright owner. All rights are reserved under United States copyright law unless specified otherwise. File name: Supplementary Information Description: Supplementary figures, supplementary tables, supplementary notes, supplementary methods and supplementary references. CCNE1 CCNE1 CCNE1 CCNE1 36 40 32 34 32 35 30 32 28 30 30 28 28 26 24 25 Normalized Expression Normalized Expression Normalized Expression Normalized Expression 26 G1 S G2/M G1 S G2/M G1 S G2/M G1 S G2/M Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage CCNE1 CCNE1 CCNE1 CCNE1 40 32 40 40 35 30 38 30 30 28 36 25 26 20 20 34 Normalized Expression Normalized Expression Normalized Expression 24 Normalized Expression G1 S G2/M G1 S G2/M G1 S G2/M G1 S G2/M Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Cell Cycle Stage Supplementary Figure 1 | High stochasticity of single-cell gene expression means, as demonstrated by relative expression levels of gene Ccne1 using the mESC-SMARTer data. For every panel, 20 sample cells were randomly selected for each of the three stages, followed by plotting the mean expression levels at each stage. -

S41467-020-18249-3.Pdf
ARTICLE https://doi.org/10.1038/s41467-020-18249-3 OPEN Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain Lei Zhao1,2,17, Zhongqi Li 1,2,17, Joaquim S. L. Vong2,3,17, Xinyi Chen1,2, Hei-Ming Lai1,2,4,5,6, Leo Y. C. Yan1,2, Junzhe Huang1,2, Samuel K. H. Sy1,2,7, Xiaoyu Tian 8, Yu Huang 8, Ho Yin Edwin Chan5,9, Hon-Cheong So6,8, ✉ ✉ Wai-Lung Ng 10, Yamei Tang11, Wei-Jye Lin12,13, Vincent C. T. Mok1,5,6,14,15 &HoKo 1,2,4,5,6,8,14,16 1234567890():,; The molecular signatures of cells in the brain have been revealed in unprecedented detail, yet the ageing-associated genome-wide expression changes that may contribute to neurovas- cular dysfunction in neurodegenerative diseases remain elusive. Here, we report zonation- dependent transcriptomic changes in aged mouse brain endothelial cells (ECs), which pro- minently implicate altered immune/cytokine signaling in ECs of all vascular segments, and functional changes impacting the blood–brain barrier (BBB) and glucose/energy metabolism especially in capillary ECs (capECs). An overrepresentation of Alzheimer disease (AD) GWAS genes is evident among the human orthologs of the differentially expressed genes of aged capECs, while comparative analysis revealed a subset of concordantly downregulated, functionally important genes in human AD brains. Treatment with exenatide, a glucagon-like peptide-1 receptor agonist, strongly reverses aged mouse brain EC transcriptomic changes and BBB leakage, with associated attenuation of microglial priming. We thus revealed tran- scriptomic alterations underlying brain EC ageing that are complex yet pharmacologically reversible. -

Methods in and Applications of the Sequencing of Short Non-Coding Rnas" (2013)
University of Pennsylvania ScholarlyCommons Publicly Accessible Penn Dissertations 2013 Methods in and Applications of the Sequencing of Short Non- Coding RNAs Paul Ryvkin University of Pennsylvania, [email protected] Follow this and additional works at: https://repository.upenn.edu/edissertations Part of the Bioinformatics Commons, Genetics Commons, and the Molecular Biology Commons Recommended Citation Ryvkin, Paul, "Methods in and Applications of the Sequencing of Short Non-Coding RNAs" (2013). Publicly Accessible Penn Dissertations. 922. https://repository.upenn.edu/edissertations/922 This paper is posted at ScholarlyCommons. https://repository.upenn.edu/edissertations/922 For more information, please contact [email protected]. Methods in and Applications of the Sequencing of Short Non-Coding RNAs Abstract Short non-coding RNAs are important for all domains of life. With the advent of modern molecular biology their applicability to medicine has become apparent in settings ranging from diagonistic biomarkers to therapeutics and fields angingr from oncology to neurology. In addition, a critical, recent technological development is high-throughput sequencing of nucleic acids. The convergence of modern biotechnology with developments in RNA biology presents opportunities in both basic research and medical settings. Here I present two novel methods for leveraging high-throughput sequencing in the study of short non- coding RNAs, as well as a study in which they are applied to Alzheimer's Disease (AD). The computational methods presented here include High-throughput Annotation of Modified Ribonucleotides (HAMR), which enables researchers to detect post-transcriptional covalent modifications ot RNAs in a high-throughput manner. In addition, I describe Classification of RNAs by Analysis of Length (CoRAL), a computational method that allows researchers to characterize the pathways responsible for short non-coding RNA biogenesis. -

Genomic and Transcriptomic Investigations Into the Feed Efficiency Phenotype of Beef Cattle
Provided by the author(s) and NUI Galway in accordance with publisher policies. Please cite the published version when available. Title Genomic and transcriptomic investigations into the feed efficiency phenotype of beef cattle Author(s) Higgins, Marc Publication Date 2019-03-06 Publisher NUI Galway Item record http://hdl.handle.net/10379/15008 Downloaded 2021-09-25T18:07:39Z Some rights reserved. For more information, please see the item record link above. Genomic and Transcriptomic Investigations into the Feed Efficiency Phenotype of Beef Cattle Marc Higgins, B.Sc., M.Sc. A thesis submitted for the Degree of Doctor of Philosophy to the Discipline of Biochemistry, School of Natural Sciences, National University of Ireland, Galway. Supervisor: Dr. Derek Morris Discipline of Biochemistry, School of Natural Sciences, National University of Ireland, Galway. Supervisor: Dr. Sinéad Waters Teagasc, Animal and Bioscience Research Department, Animal & Grassland Research and Innovation Centre, Teagasc, Grange. Submitted November 2018 Table of Contents Declaration ................................................................................................................ vii Funding .................................................................................................................... viii Acknowledgements .................................................................................................... ix Abstract ...................................................................................................................... -

Alternative Splicing in the Nuclear Receptor Superfamily Expands Gene Function to Refine Endo-Xenobiotic Metabolism S
Supplemental material to this article can be found at: http://dmd.aspetjournals.org/content/suppl/2020/01/24/dmd.119.089102.DC1 1521-009X/48/4/272–287$35.00 https://doi.org/10.1124/dmd.119.089102 DRUG METABOLISM AND DISPOSITION Drug Metab Dispos 48:272–287, April 2020 Copyright ª 2020 by The American Society for Pharmacology and Experimental Therapeutics Minireview Alternative Splicing in the Nuclear Receptor Superfamily Expands Gene Function to Refine Endo-Xenobiotic Metabolism s Andrew J. Annalora, Craig B. Marcus, and Patrick L. Iversen Department of Environmental and Molecular Toxicology, Oregon State University, Corvallis, Oregon (A.J.A., C.B.M., P.L.I.) and United States Army Research Institute for Infectious Disease, Frederick, Maryland (P.L.I.) Received August 16, 2019; accepted December 31, 2019 ABSTRACT Downloaded from The human genome encodes 48 nuclear receptor (NR) genes, whose Exon inclusion options are differentially distributed across NR translated products transform chemical signals from endo- subfamilies, suggesting group-specific conservation of resilient func- xenobiotics into pleotropic RNA transcriptional profiles that refine tionalities. A deeper understanding of this transcriptional plasticity drug metabolism. This review describes the remarkable diversifica- expands our understanding of how chemical signals are refined and tion of the 48 human NR genes, which are potentially processed into mediated by NR genes. This expanded view of the NR transcriptome dmd.aspetjournals.org over 1000 distinct mRNA transcripts by alternative splicing (AS). The informs new models of chemical toxicity, disease diagnostics, and average human NR expresses ∼21 transcripts per gene and is precision-based approaches to personalized medicine. -

An Ultraconserved Element Controls Homeostatic Splicing of ARGLU1 Mrna Stephan P
University of Connecticut OpenCommons@UConn Doctoral Dissertations University of Connecticut Graduate School 4-26-2017 An Ultraconserved Element Controls Homeostatic Splicing of ARGLU1 mRNA Stephan P. Pirnie University of Connecticut School of Medicine, [email protected] Follow this and additional works at: https://opencommons.uconn.edu/dissertations Recommended Citation Pirnie, Stephan P., "An Ultraconserved Element Controls Homeostatic Splicing of ARGLU1 mRNA" (2017). Doctoral Dissertations. 1428. https://opencommons.uconn.edu/dissertations/1428 An Ultraconserved Element Controls Homeostatic Splicing of ARGLU1 mRNA Stephan P Pirnie, PhD University of Connecticut, 2017 Abstract ARGLU1 is a well conserved protein whose function is not well understood. I will show that ARGLU1 is an alternatively spliced gene, with at least three alternatively spliced isoforms. The main isoform codes for a nuclear protein that has been associated with the mediator transcriptional regulation complex as well as components of the spliceosome. One alternative isoform of ARGLU1 retains a single unspliced intron, even though all other introns have been removed, and is localized exclusively in the nucleus. A second alternative isoform causes inclusion of a premature termination codon, and is quickly degraded by the nonsense mediated decay quality control pathway. Interestingly, the retained intron contains an ultraconserved element, which is more than 95% conserved between human and chicken for over 500 bases. I will show that this ultraconserved element plays a key role in the alternative splicing of ARGLU1. Furthermore, I will show that exogenous overexpression of ARGLU1 leads to dramatic changes in alternative splicing of its own endogenous mRNA, causing a decrease in the protein coding isoform, and an increase in the retained intron and nonsense mediated decay targeted isoforms. -

Supplementary Table 1 Double Treatment Vs Single Treatment
Supplementary table 1 Double treatment vs single treatment Probe ID Symbol Gene name P value Fold change TC0500007292.hg.1 NIM1K NIM1 serine/threonine protein kinase 1.05E-04 5.02 HTA2-neg-47424007_st NA NA 3.44E-03 4.11 HTA2-pos-3475282_st NA NA 3.30E-03 3.24 TC0X00007013.hg.1 MPC1L mitochondrial pyruvate carrier 1-like 5.22E-03 3.21 TC0200010447.hg.1 CASP8 caspase 8, apoptosis-related cysteine peptidase 3.54E-03 2.46 TC0400008390.hg.1 LRIT3 leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 1.86E-03 2.41 TC1700011905.hg.1 DNAH17 dynein, axonemal, heavy chain 17 1.81E-04 2.40 TC0600012064.hg.1 GCM1 glial cells missing homolog 1 (Drosophila) 2.81E-03 2.39 TC0100015789.hg.1 POGZ Transcript Identified by AceView, Entrez Gene ID(s) 23126 3.64E-04 2.38 TC1300010039.hg.1 NEK5 NIMA-related kinase 5 3.39E-03 2.36 TC0900008222.hg.1 STX17 syntaxin 17 1.08E-03 2.29 TC1700012355.hg.1 KRBA2 KRAB-A domain containing 2 5.98E-03 2.28 HTA2-neg-47424044_st NA NA 5.94E-03 2.24 HTA2-neg-47424360_st NA NA 2.12E-03 2.22 TC0800010802.hg.1 C8orf89 chromosome 8 open reading frame 89 6.51E-04 2.20 TC1500010745.hg.1 POLR2M polymerase (RNA) II (DNA directed) polypeptide M 5.19E-03 2.20 TC1500007409.hg.1 GCNT3 glucosaminyl (N-acetyl) transferase 3, mucin type 6.48E-03 2.17 TC2200007132.hg.1 RFPL3 ret finger protein-like 3 5.91E-05 2.17 HTA2-neg-47424024_st NA NA 2.45E-03 2.16 TC0200010474.hg.1 KIAA2012 KIAA2012 5.20E-03 2.16 TC1100007216.hg.1 PRRG4 proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) 7.43E-03 2.15 TC0400012977.hg.1 SH3D19 -
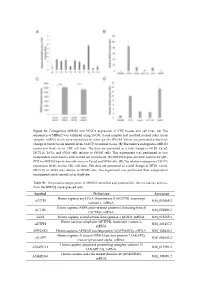
Figure S1. Endogenous MIR45
Figure S1. Endogenous MIR452 and VEGFA expression in CRC tissues and cell lines. (A) The expression of MIR452 was validated using 10 CRC tissue samples and matched normal colon tissue samples. miRNA levels were normalized to colon-specific RNU48. Values are presented as the fold- change in tumor tissue relative levels (ΔΔCT) to normal tissue. (B) The relative endogenous MIR452 expression levels in six CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, LoVo, and SW48 cells relative to SW480 cells. This experiment was performed as two independent experiments, each carried out in triplicate. (C) MIR452 expression level analysis by qRT- PCR for MIR452 transfection efficiency in Caco2 and SW48 cells. (D) The relative endogenous VEGFA expression levels in five CRC cell lines. The data are presented as a fold change in HT29, Caco2, HCT116, or SW48 cells relative to SW480 cells. This experiment was performed three independent experiments, each carried out in duplicate. Table S1. The putative target genes of MIR452 identified and predicted by the microarray analysis from the MIR452 overexpressed cells. Symbol Definition Accession Homo sapiens acyl-CoA thioesterase 8 (ACOT8), transcript ACOT8 NM_005469.2 variant 1, mRNA. Homo sapiens ARP6 actin-related protein 6 homolog (yeast) ACTR6 NM_022496.3 (ACTR6), mRNA. ADI1 Homo sapiens acireductone dioxygenase 1 (ADI1), mRNA. NM_018269.1 Homo sapiens aftiphilin (AFTPH), transcript variant 1, AFTPH NM_203437.2 mRNA. AHNAK2 Homo sapiens AHNAK nucleoprotein 2 (AHNAK2), mRNA. NM_138420.2 Homo sapiens A kinase (PRKA) anchor protein 7 (AKAP7), AKAP7 NM_004842.2 transcript variant alpha, mRNA. Homo sapiens anaphase promoting complex subunit 13 ANAPC13 NM_015391.2 (ANAPC13), mRNA. -

The Neurodegenerative Diseases ALS and SMA Are Linked at The
Nucleic Acids Research, 2019 1 doi: 10.1093/nar/gky1093 The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex Downloaded from https://academic.oup.com/nar/advance-article-abstract/doi/10.1093/nar/gky1093/5162471 by [email protected] on 06 November 2018 Binkai Chi, Jeremy D. O’Connell, Alexander D. Iocolano, Jordan A. Coady, Yong Yu, Jaya Gangopadhyay, Steven P. Gygi and Robin Reed* Department of Cell Biology, Harvard Medical School, 240 Longwood Ave. Boston MA 02115, USA Received July 17, 2018; Revised October 16, 2018; Editorial Decision October 18, 2018; Accepted October 19, 2018 ABSTRACT Fused in Sarcoma (FUS) and TAR DNA Binding Protein (TARDBP) (9–13). FUS is one of the three members of Understanding the molecular pathways disrupted in the structurally related FET (FUS, EWSR1 and TAF15) motor neuron diseases is urgently needed. Here, we family of RNA/DNA binding proteins (14). In addition to employed CRISPR knockout (KO) to investigate the the RNA/DNA binding domains, the FET proteins also functions of four ALS-causative RNA/DNA binding contain low-complexity domains, and these domains are proteins (FUS, EWSR1, TAF15 and MATR3) within the thought to be involved in ALS pathogenesis (5,15). In light RNAP II/U1 snRNP machinery. We found that each of of the discovery that mutations in FUS are ALS-causative, these structurally related proteins has distinct roles several groups carried out studies to determine whether the with FUS KO resulting in loss of U1 snRNP and the other two members of the FET family, TATA-Box Bind- SMN complex, EWSR1 KO causing dissociation of ing Protein Associated Factor 15 (TAF15) and EWS RNA the tRNA ligase complex, and TAF15 KO resulting in Binding Protein 1 (EWSR1), have a role in ALS. -

Anti-ARGLU1 Antibody (ARG40881)
Product datasheet [email protected] ARG40881 Package: 100 μl anti-ARGLU1 antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes ARGLU1 Tested Reactivity Hu Predict Reactivity Bov Tested Application FACS, IHC-P, WB Host Rabbit Clonality Polyclonal Isotype IgG Target Name ARGLU1 Antigen Species Human Immunogen KLH-conjugated synthetic peptide corresponding to aa. 48-74 of Human ARGLU1. Conjugation Un-conjugated Alternate Names Arginine and glutamate-rich protein 1 Application Instructions Application table Application Dilution FACS 1:10 - 1:50 IHC-P 1:50 - 1:100 WB 1:1000 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 33 kDa Observed Size ~ 38 kDa Properties Form Liquid Purification Purification with Protein A and immunogen peptide. Buffer PBS and 0.09% (W/V) Sodium azide. Preservative 0.09% (W/V) Sodium azide. Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C or below. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. www.arigobio.com 1/2 Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol ARGLU1 Gene Full Name arginine and glutamate rich 1 Function Required for the estrogen-dependent expression of ESR1 target genes. Can act in cooperation with MED1. [UniProt] Cellular Localization Nucleus. Note=Recruited, in an estrogen-dependent manner, to ESR1 target gene promoters.