OMONTYS® Safely and Effectively
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Specialty Drug List 10-22-12 Final
Iowa Medicaid Specialty Drug List Effective 10/22/2012 “Specialty” drugs include biological drugs, blood-derived products, complex molecules, and select oral, injectable, and infused medications identified by the Department and reimbursed at AWP-17% plus the dispensing fee. Specialty pricing will be applied to both the brand and generic drug products. NOTE: See the PDL at www.iowamedicaidpdl.com for specific PA criteria for the following drugs. BRAND NAME GENERIC NAME AGENTS FOR GAUCHER DISEASE ELELYSO taliglucerase alfa VPRIV velaglucerase alfa ALS RILUTEK riluzole ALCOHOL DEPENDENCE VIVITROL naltrexone AMINOGLYCOSIDES TOBI tobramycin ANTI-ASTHMATICS ALPHA PROTEINASE INHIBITORS ARALAST proteinase inhibitor ARALAST NP proteinase inhibitor PROLASTIN, C proteinase inhibitor ZEMAIRA alpha-1 proteinase inhibitor ANTIASTHMATIC - BETA - ADRENERGICS BRETHINE INJECTION turbutaline sulfate ANTIASTHMATIC - HYDRO-LYTIC ENZYMES KALYDECO ivacaftor ANTIBIOTICS - MISC. CAYSTON aztreonam lysine for inhal soln ANTI-CATAPLECTIC AGENTS XYREM sodium oxybate oral solution ANTICOAGULANTS ARIXTRA fondaparinux sodium FRAGMIN dalteparin sodium INNOHEP tinzaparin sodium LOVENOX enoxaparin sodium ANTICONVULSANTS SABRIL vigabatrin ANTIDOTES FERRIPROX deferiprone ANTIDOTES - CHELATING AGENTS CHEMET succimer EXJADE deferasirox tab for oral susp Page 1 of 10 ANTIEMETIC - TETRAHYDROCANNABINOL (THC) DERIVATIVES MARINOL dronabinol ANTIFUNGALS - ASSORTED AMBISOME amphotericin B Liposome IV for suspension ANCOBON flucytosine CANCIDAS caspofungin acetate for IV solution -

DRUGS REQUIRING PRIOR AUTHORIZATION in the MEDICAL BENEFIT Page 1
Effective Date: 08/01/2021 DRUGS REQUIRING PRIOR AUTHORIZATION IN THE MEDICAL BENEFIT Page 1 Therapeutic Category Drug Class Trade Name Generic Name HCPCS Procedure Code HCPCS Procedure Code Description Anti-infectives Antiretrovirals, HIV CABENUVA cabotegravir-rilpivirine C9077 Injection, cabotegravir and rilpivirine, 2mg/3mg Antithrombotic Agents von Willebrand Factor-Directed Antibody CABLIVI caplacizumab-yhdp C9047 Injection, caplacizumab-yhdp, 1 mg Cardiology Antilipemic EVKEEZA evinacumab-dgnb C9079 Injection, evinacumab-dgnb, 5 mg Cardiology Hemostatic Agent BERINERT c1 esterase J0597 Injection, C1 esterase inhibitor (human), Berinert, 10 units Cardiology Hemostatic Agent CINRYZE c1 esterase J0598 Injection, C1 esterase inhibitor (human), Cinryze, 10 units Cardiology Hemostatic Agent FIRAZYR icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent HAEGARDA c1 esterase J0599 Injection, C1 esterase inhibitor (human), (Haegarda), 10 units Cardiology Hemostatic Agent ICATIBANT (generic) icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent KALBITOR ecallantide J1290 Injection, ecallantide, 1 mg Cardiology Hemostatic Agent RUCONEST c1 esterase J0596 Injection, C1 esterase inhibitor (recombinant), Ruconest, 10 units Injection, lanadelumab-flyo, 1 mg (code may be used for Medicare when drug administered under Cardiology Hemostatic Agent TAKHZYRO lanadelumab-flyo J0593 direct supervision of a physician, not for use when drug is self-administered) Cardiology Pulmonary Arterial Hypertension EPOPROSTENOL (generic) -

Freedom of Information Act 2000
FREEDOM OF INFORMATION ACT 2000 THE ROYAL CORNWALL HOSPITALS NHS TRUST RESPONSE TO INFORMATION REQUEST Date Request Received: 15th November 2019 FOI Ref: 8454 Requested Information Question for your pharmacy and/or procurement team regarding the number of medicines and/or nursing services provided to NHS patients by an Independent Homecare Provider 1) In your organisation, which named individuals have the overall responsibility for any homecare provision for your patients? 2) Do you currently have in post an operational lead for homecare services in your organisation – If so, what is their name/role? 3) What are your organisations minimum requirements for accepting a homecare provider? 4) If you have an outsourced outpatient pharmacy, are they able to provide nurse services / training for patients on how to self-inject for medicines administered by sub-cutaneous injection as part of their contract? Can you please advise of total numbers of NHS patients who; 5) Received a homecare delivery service of drug and/or nurse service at dates Jan 2018 / Jan 2019 / October 2019 – please provide these numbers by; a) Drug name b) Therapy / clinical area c) Name of the homecare provider who provided/provides this service d) If possible please identify if these services are NHS funded or pharmaceutical / manufacturer funded services. Response 1) The Chief Pharmacist at the Royal Cornwall Hospitals Trust has overall responsibility for Medicines Homecare services. 2) The Royal Cornwall Hospitals Trust Chief Pharmacy Technician is the Operational Lead -

Biosimilar Epoetins and Other ``Follow-On
Biosimilar epoetins and other “follow-on” biologics: Update on the European experiences Wolfgang Jelkmann To cite this version: Wolfgang Jelkmann. Biosimilar epoetins and other “follow-on” biologics: Update on the European experiences. American Journal of Hematology, Wiley, 2010, 85 (10), pp.771. 10.1002/ajh.21805. hal-00552331 HAL Id: hal-00552331 https://hal.archives-ouvertes.fr/hal-00552331 Submitted on 6 Jan 2011 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. American Journal of Hematology Biosimilar epoetins and other “follow-on” biologics: Update on the European experiences For Peer Review Journal: American Journal of Hematology Manuscript ID: AJH-10-0229.R1 Wiley - Manuscript type: Critical Review Date Submitted by the 10-Jun-2010 Author: Complete List of Authors: Jelkmann, Wolfgang; University, Physiology Anemias, Erythropoietin, Hematology- medical, Neutropenia, Keywords: Pharmacology John Wiley & Sons Page 1 of 30 American Journal of Hematology 1 2 3 Table II. Benefits and problems related to the use of biosimilars 4 5 ________________________________________________________________ 6 Benefits Problems 7 ______________________________________________________________________ 8 9 10 Lower pricing than originator medicines Lack of long-term experience 11 (efficacy, safety, immunogenicity?) 12 13 Pressure on innovator companies Product-specific administration routes 14 15 to reduce prices of originator medicines (s.c. -

Myelodysplastic Syndromes
MYELODYSPLASTIC SYNDROMES TREATMENT REGIMENS (Part 1 of 3) Clinical Trials: The National Comprehensive Cancer Network recommends cancer patient participation in clinical trials as the gold standard for treatment. Cancer therapy selection, dosing, administration, and the management of related adverse events can be a complex process that should be handled by an experienced healthcare team. Clinicians must choose and verify treatment options based on the individual patient; drug dose modifications and supportive care interventions should be administered accordingly. The cancer treatment regimens below may include both U.S. Food and Drug Administration-approved and unapproved indications/regimens. These regimens are only provided to supplement the latest treatment strategies. These Guidelines are a work in progress that may be refined as often as new significant data becomes available. The NCCN Guidelines® are a consensus statement of its authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult any NCCN Guidelines® is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any way. First-Line Treatment1 Note: All recommendations are Category 2A unless otherwise indicated. Relatively Lower-Risk Patientsa,b Symptomatic Anemia With del(5q) ± One Other Cytogenetic Abnormality (except those involving chromosome 7) REGIMEN DOSING Lenalidomide2-5,c Days 1–21: Lenalidomide 10mg orally once daily. Repeat cycle every 28 days (or 28 days monthly). Assess response 2–4 months after initiation of treatment. -

Erythropoiesis-Stimulating Agents (Esas) C15389-A
Drug and Biologic Coverage Criteria Effective Date: 08/01/2017 Last P&T Approval/Version: 07/28/2021 Next Review Due By: 08/2022 Policy Number: C15389-A Erythropoiesis-stimulating agents (ESAs) PRODUCTS AFFECTED Aranesp (darbepoetin alfa), Epogen (epoetin alfa), Procrit (epoetin alfa), RETACRIT™ (epoetin alfa- epbx), Mircera'" (methoxy polyethylene glycol-epoetin beta) COVERAGE POLICY Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide Molina Healthcare complete medical rationale when requesting any exceptions to these guidelines Documentation Requirements: Molina Healthcare reserves the right to require that additional documentation be made available as part of its coverage determination; quality improvement; and fraud; waste and abuse prevention processes. Documentation required may include, but is not limited to, patient records, test results and credentials of the provider ordering or performing a drug or service. Molina Healthcare may deny reimbursement or take additional appropriate action if the documentation provided does not support the initial -

Specialty Drug Benefit Document
Louisiana Healthcare Connections Specialty Drug Benefit ouisiana Healthcare Connections provides coverage of a number of specialty drugs. All specialty drugs, such as biopharmaceuticals and injectables, require a prior authorization (PA) to be approved for L payment by Louisiana Healthcare Connections. PA requirements are programmed specific to the drug. Since the list of specialty drugs changes over time due to new drug arrivals and other market conditions, it is important to contact Provider Services at 1-866-595-8133 or check the Louisiana Healthcare Connections website at www.LouisianaHealthConnect.com for updates to this benefit. Requests for specialty drugs can be submitted to Louisiana Healthcare Connections by filling out the Medication Prior Authorization Form that is available on the Louisiana Healthcare Connections website at www.LouisianaHealthConnect.com and faxing the request as instructed on the form. Louisiana Healthcare Connections members can receive the specialty drugs they require at any outpatient pharmacy enrolled in our pharmacy network that can supply specialty drugs. Providers that wish to have drugs distributed by a SPECIALTY PHARMACY should FAX the request to 1-866-399-0929 for review. If a provider wishes to dispense a specialty drug from OFFICE STOCK, the provider should FAX the request to Louisiana Healthcare Connections at 1-877-401-8172 for review. BRAND NAME INGREDIENTS SPECIAL INSTRUCTIONS ACTEMRA TOCILIZUMAB ACTHAR HP CORTICOTROPIN ACTIMMUNE INTERFERON GAMMA-1B ADAGEN PEGADEMASE BOVINE Limited Distribution -

MSM Chapter 1200 3/1/21
MEDICAID SERVICES MANUAL TRANSMITTAL LETTER February 23, 2021 TO: CUSTODIANS OF MEDICAID SERVICES MANUAL FROM: JESSICA KEMMERER, HIPAA PRIVACY AND CIVIL RIGHTS OFFICER /Jessica Kemmerer/ BACKGROUND AND EXPLANATION The DHCFP is proposing revisions to Medicaid Services Manual (MSM), Chapter 1200 – Prescribed Drugs, Appendix A, to reflect recommendations approved on October 22, 2020, by the Drug Use Review (DUR) Board. The proposed changes include the addition of new prior authorization criteria for Doxepine Topical, the addition of new prior authorization criteria for Zeposia® (ozanimod), addition of new prior authorization for Evenity® (romosozumab-aqqg), Prolia® (denosumab), Forteo® (teriparatide) and Tymlos® (abaloparatide) within a new combined osteoporosis agents section, and addition of new prior authorization criteria for Orilissa® (elagolix) and Oriahnn® (elagolix, estradiol, and norethindrone) within a new Gonadorpin Hormone Receptor (GnRH) Antagonist and Combinations section. Additionally, the DHCFP is proposing revisions to the existing prior authorization criteria for psychotropic medications for children and adolescents, and revision to the existing clinical criteria for Epidiolex® (cannabidiol). Throughout the chapter, grammar, punctuation and capitalization changes were made, duplications removed, acronyms used and standardized, and language reworded for clarity. Renumbering and re- arranging of sections was necessary. These changes are effective March 1, 2021. MATERIAL TRANSMITTED MATERIAL SUPERSEDED MTL N/A MTL N/A MSM Ch 1200 – Prescribed Drugs MSM Ch 1200 – Prescribed Drugs Background and Explanation of Policy Changes, Manual Section Section Title Clarifications and Updates Appendix A Psychotropic Added new policy language criteria on which specific Section N Medications for drug classes may bypass polypharmacy clinical criteria. Children and Adolescents Appendix A Reserved for Future Created a new section titled “Doxepin Topical.” Added Section W Use new prior authorization criteria for doxepin topical. -
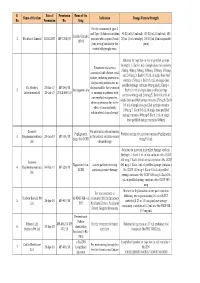
Final List of R-DNA Based Drugs Approved in the Country.Xlsx
S. Date of Permission Name of the Name of the firm Indication Dosage Form & Strength No. Permission No. Drug For the treatment of type -I and Type -II diabetes mellitus 40 IU/ml (10 ml vial), 100 IU/ml (10 ml vial), 100 Insulin Glargine 1 Wockhardt Limited 22.02.2007 MF-7206/07 patients who required basal IU/ml (3 ml cartridge), 100 IU/ml (3 ml disposable 100 IU (long acting) insulin for the pens) control of hyperglycemia. Solution for injection in vial or prefilled syringe Strength: 1. Each 1 mL of single dose vial contains Treatment of anaemia 25mcg, 40mcg, 60mcg, 100mcg, 200mcg, 300mcg associated with chronic renal and 500mcg 2. Each 0.75 mL of single dose vial failure, including patients on contains 150mcg 3. Each 0.3 mL of single dose dialysis and patients not on prefilled syringe contains 60mcg and 150mcg 4. Dr. Reddy's 23-Mar-10 MF-246/10 dialysis and for the treatment 2 Darbepoetin alfa Each 0.4 mL of single dose prefilled syringe Laboratories Ltd. 20-Jul-10 BULK-668/10 of anaemia in patients with contains 40mcg and 200mcg 5. Each 0.42 mL of non-myeloid malignancies, single dose prefilled syringe contains 25mcg 6. Each where anaemia is due to the 0.5 mL of single dose prefilled syringe contains effect of concomitantly 100mcg 7. Each 0.6 mL of single dose prefilled administered chemotherapy syringe contains 300mcg 8. Each 1 mL of single dose prefilled syringe contains 500mcg Gennova For prevention of neutropenia Pegfilgrastim Prefilled syringe for injection contains Pegfilgrastim 3 Biopharmaceuticals 29-Jan-10 MF-104/10 in the patient receiving cancer (peg-r-hu-GCSF) 6 mg/0.6 mL Ltd. -

Overview of Antibody Drug Delivery
pharmaceutics Review Overview of Antibody Drug Delivery Sahar Awwad 1,2,* ID and Ukrit Angkawinitwong 1 1 UCL School of Pharmacy, London WC1N 1AX, UK; [email protected] 2 National Institute for Health Research (NIHR) Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, London EC1 V9EL, UK * Correspondence: [email protected]; Tel.: +44-207-753-5802 Received: 27 March 2018; Accepted: 29 June 2018; Published: 4 July 2018 Abstract: Monoclonal antibodies (mAbs) are one of the most important classes of therapeutic proteins, which are used to treat a wide number of diseases (e.g., oncology, inflammation and autoimmune diseases). Monoclonal antibody technologies are continuing to evolve to develop medicines with increasingly improved safety profiles, with the identification of new drug targets being one key barrier for new antibody development. There are many opportunities for developing antibody formulations for better patient compliance, cost savings and lifecycle management, e.g., subcutaneous formulations. However, mAb-based medicines also have limitations that impact their clinical use; the most prominent challenges are their short pharmacokinetic properties and stability issues during manufacturing, transport and storage that can lead to aggregation and protein denaturation. The development of long acting protein formulations must maintain protein stability and be able to deliver a large enough dose over a prolonged period. Many strategies are being pursued to improve the formulation and dosage forms of antibodies to improve efficacy and to increase the range of applications for the clinical use of mAbs. Keywords: antibodies; protein; pharmacokinetics; drug delivery; stability 1. -

Presentation Title
The COMMANDS trial: a phase 3 study of the efficacy and safety of luspatercept versus epoetin alfa for the treatment of anemia due to Revised International Prognostic Scoring System Very Low-, Low-, or Intermediate-risk myelodysplastic syndromes in erythropoiesis stimulating agent-naive patients who require red blood cell transfusions Matteo Della Porta,1,2 Uwe Platzbecker,3 Valeria Santini,4 Guillermo Garcia-Manero,5 Rami S. Komrokji,6 Rodrigo Ito,7 Pierre Fenaux8 1Cancer Center IRCCS Humanitas Research Hospital, Milan, Italy; 2Department of Biomedical Sciences, Humanitas University, Milan, Italy; 3Medical Clinic and Policlinic 1, Hematology and Cellular Therapy, University Hospital Leipzig, Leipzig, Germany; 4Azienda Ospedaliero-Universitaria Careggi, University of Florence, Florence, Italy; 5Department of Leukemia, University of Texas MD Anderson Cancer Center, Houston, TX; 6Moffitt Cancer Center, Tampa, FL; 7Bristol Myers Squibb, Princeton, NJ; 8Service d'Hématologie Séniors, Hôpital Saint-Louis, Université Paris 7, Paris, France Presentation 2198 Presenting author disclosures M.D.P.: no conflicts of interest to disclose. 2 Introduction and objectives Introduction • Studies of epoetin alfa and darbepoetin alfa have demonstrated efficacy among patients with LR-MDS, but the patient population in which a clinically significant effect is observed may be limited1,2 • Luspatercept, a first-in-class erythroid maturation agent with a mechanism of action distinct from ESAs,3 is approved by the US FDA for the treatment of anemia failing an -

Peginesatide for Anaemia in Chronic Kidney Disease – First and Second Line
Peginesatide for anaemia in chronic kidney disease – first and second line June 2012 This technology summary is based on information available at the time of research and a limited literature search. It is not intended to be a definitive statement on the safety, efficacy or effectiveness of the health technology covered and should not be used for commercial purposes. The NIHR Horizon Scanning Centre Research Programme is part of the National Institute for Health Research www.nhsc-healthhorizons.org.uk June 2012 Peginesatide for anaemia in chronic kidney disease – first and second line Target group • Symptomatic anaemia: patients on dialysis with chronic kidney disease (CKD) – first and second line. Background Human erythropoietin (EPO) is a protein produced by the kidneys that stimulates the production of red blood cells. Because the kidney is the sole source of EPO synthesis in adults, the reduction in kidney mass that occurs in progressive CKD often causes impairment of EPO production, resulting in anaemia. Technology description Peginesatide (AF-37702) is a synthetic peptide mimetic of EPO that binds directly to the erythropoietin receptor on red blood cell precursors to stimulate red cell formation. Peginesatide is intended as a substitute for currently available erythropoiesis stimulating agents (ESAs) in the treatment of anaemia in CKD dialysis patients. It is administered intravenously (IV) or subcutaneously (SC) at 0.04 mg/kg once monthly. After the initial dose, doses may be titrated to achieve a haemoglobin (Hb) level in the range of 10 to 12g/dL. Peginesatide is also in phase II clinical trials for pure red cell aplasia.