Comparative Analysis of 37 Acinetobacter Bacteriophages
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Gut Microbiome Alterations in Ulcerative Colitis and After Moxibustion Intervention
Gut Microbiome Alterations In Ulcerative Colitis And After Moxibustion Intervention Qin Qi Shanghai University of Traditional Chinese Medicine Ya-Nan Liu Shanghai University of Traditional Chinese Medicine Si-Yi Lv Shanghai University of Traditional Chinese Medicine Huan-Gan Wu Shanghai University of Traditional Chinese Medicine Lin-Shuang Zhang Zhejiang Institute for Food and Drug Control Zhan Cao Tongji University School of Medicine Hui-Rong Liu Shanghai University of Traditional Chinese Medicine Xiao-Mei Wang ( [email protected] ) Shanghai University of Traditional Chinese Medicine Lu-Yi Wu Shanghai University of Traditional Chinese Medicine Research Article Keywords: Ulcerative colitis, Moxibustion, Gut microbiota, Metagenomic Posted Date: August 11th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-789670/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/22 Abstract Background: Recent studies have shown that the pathogenesis of ulcerative colitis (UC) is closely related to the gut microbiota. Moxibustion, a common treatment in traditional Chinese medicine, is the burning of the herb moxa over acupuncture points. Moxibustion has been used to improve the inammation and gastrointestinal dysfunctions in gastrointestinal disorders such as UC. In this study, we investigated whether moxibustion could improve the gut microbial dysbiosis induced by dextran sulphate sodium (DSS). Methods: Twenty-ve male rats were randomly assigned into ve groups: normal (NG), UC model (UC), moxibustion (UC+MOX), mesalazine (UC+MES), and normal rats with moxibustion (NG+MOX). The UC rat model was established by administering DSS solution. The rats in the UC+MOX and NG+MOX groups were treated with moxibustion at Tianshu (bilateral, ST25) points once daily for 7 consecutive days, and the UC+MES group rats were treated with mesalazine once daily for 7 consecutive days. -

Identification, Molecular Epidemiology, and Antibiotic Resistance Characterization of Acinetobacter Spp
FACULTY OF HEALTH SCIENCES DEPARTMENT OF MEDICAL BIOLOGY UNIVERSITY HOSPITAL OF NORTH NORWAY DEPARTMENT OF MICROBIOLOGY AND INFECTION CONTROL REFERENCE CENTRE FOR DETECTION OF ANTIMICROBIAL RESISTANCE Identification, molecular epidemiology, and antibiotic resistance characterization of Acinetobacter spp. clinical isolates Nabil Karah A dissertation for the degree of Philosophiae Doctor June 2011 Acknowledgments The work presented in this thesis has been carried out between January 2009 and September 2011 at the Reference Centre for Detection of Antimicrobial Resistance (K-res), Department of Microbiology and Infection Control, University Hospital of North Norway (UNN); and the Research Group for Host–Microbe Interactions, Department of Medical Biology, Faculty of Health Sciences, University of Tromsø (UIT), Tromsø, Norway. I would like to express my deep and truthful acknowledgment to my main supervisor Ørjan Samuelsen. His understanding and encouraging supervision played a major role in the success of every experiment of my PhD project. Dear Ørjan, I am certainly very thankful for your indispensible contribution in all the four manuscripts. I am also very grateful to your comments, suggestions, and corrections on the present thesis. I am sincerely grateful to my co-supervisor Arnfinn Sundsfjord for his important contribution not only in my MSc study and my PhD study but also in my entire career as a “Medical Microbiologist”. I would also thank you Arnfinn for your nonstop support during my stay in Tromsø at a personal level. My sincere thanks are due to co-supervisors Kristin Hegstad and Gunnar Skov Simonsen for the valuable advice, productive comments, and friendly support. I would like to thank co-authors Christian G. -

A Comparison of Genospecies of Clinical Isolates in the Acinetobacter Spp. Complex Obtained from Hospitalized Patients in Busan, Korea
Biomedical Science Letters 2019, 25(1): 40 ~53 Original Article https://doi.org/10.15616/BSL.2019.25.1.40 eISSN : 2288-7415 A Comparison of Genospecies of Clinical Isolates in the Acinetobacter spp. Complex Obtained from Hospitalized Patients in Busan, Korea Gyu-Nam Park 1,*, Hye-Sook Kang 2,** , Hye-Ran Kim 3,** *, Bo-Kyung Jung 1,*, Do-Hee Kim 4,* and Kyung-Soo Chang 1, †,*** 1Department of Clinical Laboratory Science, College of Health Sciences, Catholic University of Pusan, Busan 46252, Korea 2Department of Laboratory Medicine, Maryknoll Medical Center, Busan 48972, Korea 3Department of Clinical Laboratory Science, College of Health and Therapy, Daegu Haany University, Gyeongsangbuk-Do 38610, Korea 4Department of Laboratory Medicine, Busan Veterans Hospital, Busan 46996, Korea Of the Acinetobacter spp., A. baumannii (genospecies 2) is the most clinically significant in terms of hospital-acquired infections worldwide. It is difficult to perform Acinetobacter -related taxonomy using phenotypic characteristics and routine laboratory methods owing to clusters of closely related species. The ability to accurately identify Acinetobacter spp. is clinically important because antimicrobial susceptibility and clinical relevance differs significantly among the different genospecies. Based on the medical importance of pathogenic Acinetobacter spp., the distribution and characterization of Acinetobacter spp. isolates from 123 clinical samples was determined in the current study using four typically applied bacterial identification methods; partial rpoB gene sequencing, amplified rRNA gene restriction analysis (ARDRA) of the intergenic transcribed spacer (ITS) region of the 16 ~23S rRNA, the VITEK ® 2 system (an automated microbial identification system) and matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS). -

Detection of Acinetobacter Spp. in Blood Cultures by an Improved Fluorescent in Situ Hybridization Assay
Polish Journal of Microbiology 2018, Vol. 67, No 1, 3–10 ORIGINAL PAPER Detection of Acinetobacter spp. in Blood Cultures by an Improved Fluorescent in Situ Hybridization Assay HANIEH ASAADI1, 2, BEHROUZ NAEIMI1, 3, SOMAYYEH GHARIBI4, ABDALNASER KHOSRAVI1, SINA DOBARADARAN5, REZA TAHERKHANI1, 3 and SAEED TAJBAKHSH1, 3* 1 Department of Microbiology and Parasitology, Faculty of Medicine, Bushehr University of Medical Sciences, Bushehr, Iran 2 Student Research Committee, Bushehr University of Medical Sciences, Bushehr, Iran 3 The Persian Gulf Tropical Medicine Research Center, Bushehr University of Medical Sciences, Bushehr, Iran 4 Department of Microbiology, Faculty of Biological Sciences, Alzahra University, Tehran, Iran 5 Department of Environmental Health Engineering, Faculty of Health, Bushehr University of Medical Sciences, Bushehr, Iran Submitted 25 May 2017, revised 1 October 2017, accepted 28 November 2017 Abstract Fluorescent in situ hybridization (FISH) allows rapid detection of microorganisms. We aimed (i) to evaluate the sensitivity and specific- ity of FISH for the detection of Acinetobacter spp. in blood culture specimens and (ii) to test the simultaneous application of two genus- specific probes labeled with the same fluorochrome to increase the fluorescent signal intensity and improve the detection of Acinetobacter spp. Three hundred and twenty blood culture specimens were testedvia both the conventional laboratory methods and FISH to detect Acinetobacter spp. The specimens were examined separately with each genus-specific probe Aci and ACA, and also using a mixture of the both probes Aci and ACA. In all examinations, probe EUB338 was used accompanied by Aci and ACA. The specificity of FISH was 100% (97.5% confidence interval [CI] = 98.7% – 100%). -

The Genetic Analysis of an Acinetobacter Johnsonii Clinical Strain Evidenced the Presence of Horizontal Genetic Transfer
RESEARCH ARTICLE The Genetic Analysis of an Acinetobacter johnsonii Clinical Strain Evidenced the Presence of Horizontal Genetic Transfer Sabrina Montaña1, Sareda T. J. Schramm2, German Matías Traglia1, Kevin Chiem1,2, Gisela Parmeciano Di Noto1, Marisa Almuzara3, Claudia Barberis3, Carlos Vay3, Cecilia Quiroga1, Marcelo E. Tolmasky2, Andrés Iriarte4, María Soledad Ramírez1,2* 1 Instituto de Investigaciones en Microbiología y Parasitología Médica (IMPaM, UBA-CONICET), Buenos Aires, Argentina, 2 Department of Biological Science, California State University Fullerton, Fullerton, CA, a11111 United States of America, 3 Laboratorio de Bacteriología Clínica, Departamento de Bioquímica Clínica, Hospital de Clínicas José de San Martín, Facultad de Farmacia y Bioquímica, Buenos Aires, Argentina, 4 Departamento de Desarrollo Biotecnológico, Instituto de Higiene, Facultad de Medicina, UdelaR, Montevideo, Uruguay * [email protected] OPEN ACCESS Abstract Citation: Montaña S, Schramm STJ, Traglia GM, Chiem K, Parmeciano Di Noto G, Almuzara M, et al. Acinetobacter johnsonii rarely causes human infections. While most A. johnsonii isolates are (2016) The Genetic Analysis of an Acinetobacter β johnsonii Clinical Strain Evidenced the Presence of susceptible to virtually all antibiotics, strains harboring a variety of -lactamases have Horizontal Genetic Transfer. PLoS ONE 11(8): recently been described. An A. johnsonii Aj2199 clinical strain recovered from a hospital in e0161528. doi:10.1371/journal.pone.0161528 Buenos Aires produces PER-2 and OXA-58. We decided to delve into its genome by obtain- Editor: Ruth Hall, University of Sydney, AUSTRALIA ing the whole genome sequence of the Aj2199 strain. Genome comparison studies on Received: March 23, 2016 Aj2199 revealed 240 unique genes and a close relation to strain WJ10621, isolated from the urine of a patient in China. -
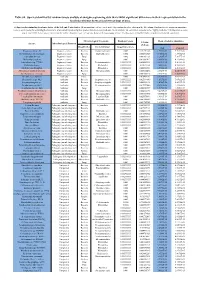
Table S8. Species Identified by Random Forests Analysis of Shotgun Sequencing Data That Exhibit Significant Differences In
Table S8. Species identified by random forests analysis of shotgun sequencing data that exhibit significant differences in their representation in the fecal microbiomes between each two groups of mice. (a) Species discriminating fecal microbiota of the Soil and Control mice. Mean importance of species identified by random forest are shown in the 5th column. Random forests assigns an importance score to each species by estimating the increase in error caused by removing that species from the set of predictors. In our analysis, we considered a species to be “highly predictive” if its importance score was at least 0.001. T-test was performed for the relative abundances of each species between the two groups of mice. P-values were at least 0.05 to be considered statistically significant. Microbiological Taxonomy Random Forests Mean of relative abundance P-Value Species Microbiological Function (T-Test) Classification Bacterial Order Importance Score Soil Control Rhodococcus sp. 2G Engineered strain Bacteria Corynebacteriales 0.002 5.73791E-05 1.9325E-05 9.3737E-06 Herminiimonas arsenitoxidans Engineered strain Bacteria Burkholderiales 0.002 0.005112829 7.1580E-05 1.3995E-05 Aspergillus ibericus Engineered strain Fungi 0.002 0.001061181 9.2368E-05 7.3057E-05 Dichomitus squalens Engineered strain Fungi 0.002 0.018887472 8.0887E-05 4.1254E-05 Acinetobacter sp. TTH0-4 Engineered strain Bacteria Pseudomonadales 0.001333333 0.025523638 2.2311E-05 8.2612E-06 Rhizobium tropici Engineered strain Bacteria Rhizobiales 0.001333333 0.02079554 7.0081E-05 4.2000E-05 Methylocystis bryophila Engineered strain Bacteria Rhizobiales 0.001333333 0.006513543 3.5401E-05 2.2044E-05 Alteromonas naphthalenivorans Engineered strain Bacteria Alteromonadales 0.001 0.000660472 2.0747E-05 4.6463E-05 Saccharomyces cerevisiae Engineered strain Fungi 0.001 0.002980726 3.9901E-05 7.3043E-05 Bacillus phage Belinda Antibiotic Phage 0.002 0.016409765 6.8789E-07 6.0681E-08 Streptomyces sp. -

Characterization of the Skin Cultivable Microbiota Composition of the Frog Pelophylax Perezi Inhabiting Different Environments
International Journal of Environmental Research and Public Health Article Characterization of the Skin Cultivable Microbiota Composition of the Frog Pelophylax perezi Inhabiting Different Environments Diogo Neves Proença 1,† , Emanuele Fasola 2,† , Isabel Lopes 2 and Paula V. Morais 1,* 1 Department of Life Sciences and Centre for Mechanical Engineering, Materials and Processes, University of Coimbra, Calçada Martim de Freitas, 3000-456 Coimbra, Portugal; [email protected] 2 CESAM and Department of Biology, University of Aveiro, 3810-005 Aveiro, Portugal; [email protected] (E.F.); [email protected] (I.L.) * Correspondence: [email protected]; Tel.: +35-1239240700 † Diogo Neves Proença and Emanuele Fasola contributed equally to this work. Abstract: Microorganisms that live in association with amphibian skin can play important roles in protecting their host. Within the scenarios of global change, it is important to understand how environmental disturbances, namely, metal pollution, can affect this microbiota. The aim of this study is to recognize core bacteria in the skin cultivable microbiota of the Perez frog (Pelophylax perezi) that are preserved regardless of the environmental conditions in which the frogs live. The characterization of these isolates revealed characteristics that can support their contributions to the ability of frogs to use metal impacted environments. Frog’s skin swabs were collected from P. perezi populations that inhabit a metal-polluted site and three reference (non-metal polluted) sites. Bacterial strains were isolated, identified, and subjected to an acid mine drainage tolerance Citation: Proença, D.N.; Fasola, E.; (AMD) test, collected upstream from a site heavily contaminated with metals, and tested to produce Lopes, I.; Morais, P.V. -

For the Analysis of Bacteriome and Resistome of Bean Sprouts
microorganisms Article Application of Nanopore Sequencing (MinION) for the Analysis of Bacteriome and Resistome of Bean Sprouts Milada Solcova * , Katerina Demnerova and Sabina Purkrtova * Department of Biochemistry and Microbiology, Faculty of Food and Biochemical Technology, University of Chemistry and Technology, Technicka 3, 16628 Prague 6, Czech Republic; [email protected] * Correspondence: [email protected] (M.S.); [email protected] (S.P.); Tel.: +420-220-44-5196 (M.S. & S.P.) Abstract: The aspiration these days is to apply rapid methods for parallel analysis of bacteriome and resistome of food samples to increase food safety and prevent antibiotic resistance genes (ARGs) spreading. In this work, we used nanopore sequencing (NS) to determine the diversity and dynamics of the microbiome and resistome in two types of bean sprouts. We proved that NS provided an easy, quick, and reliable way to identify the microbiome and resistome of a food sample also. The species diversity obtained by NS and by cultivation methods with MALDI-TOF MS identification was com- parable. In both samples, before and after cultivation (30 ◦C, 48 h), the dominant part of bacteriome formed Gammaproteobacteria (Enterobacteriaceae, Erwiniaceae, Pseudomonadaceae, Moraxellaceae) and then Firmicutes (Streptococcaceae). The diversity and abundance of single ARGs groups were comparable for both samples despite bacteriome differences. More than 50% of the detected ARGs alignments were mutations conferring resistance to aminoglycosides (16S rRNA), resistance to fluoroquinolones (gyrA, gyrB, parC, parD) and elfamycin (EF-Tu). ARGs encoding efflux pumps formed more than 30% of the Citation: Solcova, M.; Demnerova, detected alignments. Beta-lactamases were represented by many variants, but were less abundant. -
1589446757 226 5.Pdf
Journal of Global Antimicrobial Resistance 16 (2019) 59–71 Contents lists available at ScienceDirect Journal of Global Antimicrobial Resistance journal homepage: www.elsevier.com/locate/jgar Review Acinetobacter in veterinary medicine, with an emphasis on Acinetobacter baumannii a, b a a c J.H. van der Kolk *, A. Endimiani , C. Graubner , V. Gerber , V. Perreten a Swiss Institute of Equine Medicine (ISME), Department of Clinical Veterinary Medicine, Vetsuisse Faculty, University of Bern and Agroscope, Länggassstrasse 124, 3012 Bern, Switzerland b Institute for Infectious Diseases, University of Bern, Friedbühlstrasse 51, 3001 Bern, Switzerland c Institute of Veterinary Bacteriology, University of Bern, Länggassstrasse 122, 3012 Bern, Switzerland A R T I C L E I N F O A B S T R A C T Article history: Acinetobacter spp. are aerobic, rod-shaped, Gram-negative bacteria belonging to the Moraxellaceae Received 8 September 2017 family of the class Gammaproteobacteria and are considered ubiquitous organisms. Among them, Received in revised form 11 August 2018 Acinetobacter baumannii is the most clinically significant species with an extraordinary ability to Accepted 14 August 2018 accumulate antimicrobial resistance and to survive in the hospital environment. Recent reports indicate Available online 23 August 2018 that A. baumannii has also evolved into a veterinary nosocomial pathogen. Although Acinetobacter spp. can be identified to species level using matrix-assisted laser desorption/ionisation time-of-flight mass Keywords: spectrometry (MALDI-TOF/MS) coupled with an updated database, molecular techniques are still Acinetobacter baumannii necessary for genotyping and determination of clonal lineages. It appears that the majority of infections Antimicrobial resistance Dog due to A. -
Mobile Genetic Elements Associated with Blandm-1 in Acinetobacter Spp
Mobile genetic elements associated with blaNDM-1 in Acinetobacter spp. and Vibrio cholerae. by Lim Stephen Jones A thesis submitted for the degree of Doctor of Philosophy at Cardiff University 2015 i Summary NDM-producing bacteria are associated with extensive antimicrobial resistance (AMR). This thesis reports on detailed molecular analysis, including whole genome sequencing, of Acinetobacter spp. and Vibrio cholerae isolates. A study of clinical Acinetobacter baumannii isolates from India, demonstrated spread of a single strain containing blaNDM-1 but with evidence of significant genetic plasticity between isolates. A novel plasmid, pNDM-32, was fully characterised in isolate CHI-32. This contained multiple AMR genes including blaNDM-1 and the aminoglycoside methyltransferase gene armA. A repAci10 replicase gene was identified but no conjugation machinery and the plasmid could not be transferred in conjugation experiments. A single isolate of Acinetobacter bereziniae from India contained plasmid, pNDM-40-1, harbouring blaNDM-1, which was closely related to plasmids from NDM-producing Acinetobacter spp. isolated in China, and was readily transferred into Escherichia coli and Acinetobacter pittii by conjugation. Five blaNDM-1 positive Acinetobacter spp. isolated from a faecal screening study in Pakistan also included three, clonal, Acinetobacter haemolyticus isolates harbouring a similar plasmid. Three environmental V. cholerae strains from India and a blood isolate from a traveller returning to the UK from India were found to include three distantly related strains. 2 isolates of a single strain contained an IncA/C plasmid, pNDM-116-17, harbouring AMR genes including blaNDM-1. In one isolate pNDM-116-17 had become integrated into a chromosomal region containing a SXT-like element. -

Comparative Analysis of 37 Acinetobacter Bacteriophages
viruses Article Comparative Analysis of 37 Acinetobacter Bacteriophages Dann Turner 1,*, Hans-Wolfgang Ackermann 2,†, Andrew M. Kropinski 3, Rob Lavigne 4 ID , J. Mark Sutton 5 and Darren M. Reynolds 1 1 Department of Applied Sciences, Faculty of Health and Applied Sciences, University of the West of England, Coldharbour Lane, Bristol BS16 1QY, UK; [email protected] 2 Faculty of Medicine, Department of Microbiology, Immunology and Infectiology, Université Laval, Quebec, QC G1X 46, Canada 3 Departments of Food Science, Molecular and Cellular Biology; and Pathobiology, University of Guelph, Guelph, ON N1G 2W1, Canada; [email protected] 4 Laboratory of Gene Technology, KU Leuven, Kasteelpark Arenberg 21, box 2462, 3001 Leuven, Belgium; [email protected] 5 National Infections Service, Public Health England, Porton Down, Salisbury, Wiltshire SP4 0JG, UK; [email protected] * Correspondence: [email protected]; Tel.: +44-117-328-2563 † Deceased. Received: 4 December 2017; Accepted: 22 December 2017; Published: 24 December 2017 Abstract: Members of the genus Acinetobacter are ubiquitous in the environment and the multiple-drug resistant species A. baumannii is of significant clinical concern. This clinical relevance is currently driving research on bacterial viruses infecting A. baumannii, in an effort to implement phage therapy and phage-derived antimicrobials. Initially, a total of 42 Acinetobacter phage genome sequences were available in the international nucleotide sequence databases, corresponding to a total of 2.87 Mbp of sequence information and representing all three families of the order Caudovirales and a single member of the Leviviridae. A comparative bioinformatics analysis of 37 Acinetobacter phages revealed that they form six discrete clusters and two singletons based on genomic organisation and nucleotide sequence identity. -

Exploring the Nature and Diversity of Microorganisms in Healthcare and Educational Settings
University College London Nicola D’Arcy Exploring the Nature and Diversity of Microorganisms in Healthcare and Educational Settings Eastman Dental Institute Doctor of Philosophy Supervisors: Dr David A. Spratt and Professor Nigel Klein 2014 I, Nicola D’Arcy confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. Signed………………………. Date………………………… Abstract Many human populations spend approximately 90 % of their time indoors, yet relatively little is known about the microbial communities associated with indoor environments. This is despite knowledge that these microorganisms can contribute to adverse health effects, including the acquisition of healthcare-associated infections, which cause significant morbidity and mortality. The concept of the ‘indoor microbiome’ is relatively new and to date, few studies have been field-based, systematic and long-term. Hospitals in particular, are unique environments which have been shown to drive microbial evolutionary processes as they contain a different sub-set of the human population. The study of the hospital microbiome could have important implications for healthcare and infection control. This thesis explores a range of methods for investigating microorganisms in different indoor environments, including a classroom and outpatient’s waiting areas and wards in a hospital. Results show that the classroom is much more heavily contaminated in terms of total viable counts (TVCs) of bacteria recovered than the hospital environment. This was thought to be attributed to the absence of a strict cleaning regime in the classroom. High-touch items were less contaminated than other objects, likely due to them being obvious cleaning targets.