Whole-Exome Sequencing in a Japanese Pedigree Implicates A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variation Across the Human Olfactory Receptor Repertoire Alters Odor Perception
bioRxiv preprint doi: https://doi.org/10.1101/212431; this version posted November 1, 2017. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Genetic variation across the human olfactory receptor repertoire alters odor perception Casey Trimmer1,*, Andreas Keller2, Nicolle R. Murphy1, Lindsey L. Snyder1, Jason R. Willer3, Maira Nagai4,5, Nicholas Katsanis3, Leslie B. Vosshall2,6,7, Hiroaki Matsunami4,8, and Joel D. Mainland1,9 1Monell Chemical Senses Center, Philadelphia, Pennsylvania, USA 2Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, New York, USA 3Center for Human Disease Modeling, Duke University Medical Center, Durham, North Carolina, USA 4Department of Molecular Genetics and Microbiology, Duke University Medical Center, Durham, North Carolina, USA 5Department of Biochemistry, University of Sao Paulo, Sao Paulo, Brazil 6Howard Hughes Medical Institute, New York, New York, USA 7Kavli Neural Systems Institute, New York, New York, USA 8Department of Neurobiology and Duke Institute for Brain Sciences, Duke University Medical Center, Durham, North Carolina, USA 9Department of Neuroscience, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania, USA *[email protected] ABSTRACT The human olfactory receptor repertoire is characterized by an abundance of genetic variation that affects receptor response, but the perceptual effects of this variation are unclear. To address this issue, we sequenced the OR repertoire in 332 individuals and examined the relationship between genetic variation and 276 olfactory phenotypes, including the perceived intensity and pleasantness of 68 odorants at two concentrations, detection thresholds of three odorants, and general olfactory acuity. -

Sean Raspet – Molecules
1. Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Molecular weight: 240.39 g/mol Volume (cubic Angstroems): 258.88 Atoms number (non-hydrogen): 17 miLogP: 4.43 Structure: Biological Properties: Predicted Druglikenessi: GPCR ligand -0.23 Ion channel modulator -0.03 Kinase inhibitor -0.6 Nuclear receptor ligand 0.15 Protease inhibitor -0.28 Enzyme inhibitor 0.15 Commercial name: Fructaplex© IUPAC Name: 2-(3,3-dimethylcyclohexyl)-2,5,5-trimethyl-1,3-dioxane SMILES: CC1(C)CCCC(C1)C2(C)OCC(C)(C)CO2 Predicted Olfactory Receptor Activityii: OR2L13 83.715% OR1G1 82.761% OR10J5 80.569% OR2W1 78.180% OR7A2 77.696% 2. Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Molecular weight: 239.36 Volume (cubic Angstroems): 252.83 Atoms number (non-hydrogen): 17 miLogP: 4.33 Structure: Biological Properties: Predicted Druglikeness: GPCR ligand -0.6 Ion channel modulator -0.41 Kinase inhibitor -0.93 Nuclear receptor ligand -0.17 Protease inhibitor -0.39 Enzyme inhibitor 0.01 Commercial name: Sylvoxime© IUPAC Name: N-[4-(1-ethoxyethenyl)-3,3,5,5tetramethylcyclohexylidene]hydroxylamine SMILES: CCOC(=C)C1C(C)(C)CC(CC1(C)C)=NO Predicted Olfactory Receptor Activity: OR52D1 71.900% OR1G1 70.394% 0R52I2 70.392% OR52I1 70.390% OR2Y1 70.378% 3. Commercial name: Hyperflor© IUPAC Name: 2-benzyl-1,3-dioxan-5-one SMILES: O=C1COC(CC2=CC=CC=C2)OC1 Molecular weight: 192.21 g/mol Volume -

A Multi-Cancer Gene Signature Associated with Stromal Activation
A multi-cancer gene signature associated with stromal activation Sandra Orsulic, PhD Professor of OB/GYN Women’s Cancer Program Samuel Oschin Comprehensive Cancer Institute I Cedars-Sinai Medical Center 1100011010 1001000101 0101001011 Stroma/ECM Prevents Tumor Invasion Stroma/ECM Promotes Tumor Invasion Tumor Growth and Stroma Remodeling Ki67-positive dividing cells cancer cells stroma Tumor Growth and Stroma Remodeling Ki67-positive dividing cells cancer • proliferative cells • targeted by chemotherapy • genetically unstable stroma Tumor Growth and Stroma Remodeling Ki67-positive dividing cells cancer • proliferating cells • targeted by chemotherapy • genetically unstable • slow proliferating • not targeted by chemotherapy stroma • genetically stable • enhanced remodeling predicts poor outcome Expression of COL11A1 in Intratumoral Stroma COL11A1 + Fat 0% Peritumoral ~1% stroma Tumor ~50% Intratumoral 100µm stroma COL11A1 Expression in Cancers and Corresponding Normal Tissues Smooth Muscle Actin Expression in Cancers and Corresponding Normal Tissues COL11A1 Expression in Cancers and Corresponding Normal Tissues BREAST COLORECTAL 1 2 1 2 3 1. Breast (61) 1. Colon (19) 2. Invasive ductal breast 2. Rectum (3) carcinoma (389) 3. Colon adenocarcinoma (101) Overexpression gene rank: 1 Overexpression gene rank: 3 (in top 1%) (in top 1%) p-value: 1.15E-73 p-value: 2.19E-44 t-test: 33.769 t-test: 27.871 fold change: 40.542 fold change: 32.796 Increase in COL11A1 Levels During Cancer Progression Breast Cancer Ductal Invasive Carcinoma Ductal In -

Preview of “Supplement Table S4.Xls”
Annotations for all recurrently lost regions cytoband 1p36.12 3p14.2 3q13.13 q value 0,01352 0,1513 0,085307 residual q value 0,01352 0,1513 0,085307 wide peak boundaries chr1:23457835-23714048 chr3:60396160-60637030 chr3:110456383-110657226 genes in wide peak E2F2 FHIT DPPA4 ID3 DPPA2 TCEA3 DDEFL1 TBC1D19 PI4K2B STIM2 GBA3 KCNIP4 DKFZp761B107 C4orf28 GPR125 FLJ45721 hsa-mir-218-1 CXCL3 GRSF1 HNRPD HTN1 HTN3 IBSP IGFBP7 IGJ IL8 CXCL10 KDR CXCL9 AFF1 MUC7 NKX6-1 PF4 PF4V1 PKD2 POLR2B PPEF2 PPAT PPBP PRKG2 MAPK10 PTPN13 REST CXCL6 CXCL11 CXCL5 SPINK2 SPP1 SRP72 STATH SULT1E1 UGT2B4 UGT2B7 UGT2B10 UGT2B15 UGT2B17 SPARCL1 VDP SLC4A4 HERC3 GENX-3414 CDKL2 TMPRSS11D ABCG2 ADAMTS3 CLOCK CEP135 G3BP2 HNRPDL ENAM FAM13A1 CXCL13 PAICS UGT2B11 HPSE NMU SMR3B NPFFR2 UGT2A1 CCNI hsa-mir-491 hsa-mir-31 4p15.32 4q13.1 4q13.1 (continued) 6q14.1 0,14888 0,17878 0,093693 0,14888 0,17878 0,093693 chr4:17969802-29966659 chr4:55756035-90966136 chr6:76830186-107898353 CCKAR AFM SEC31A AIM1 DHX15 AFP RUFY3 BCKDHB RBPSUH ALB WDFY3 PRDM1 SOD3 AMBN LPHN3 CCNC SLIT2 ANXA3 DKFZP564O0823 CGA SLC34A2 AREG RCHY1 CNR1 PPARGC1A ART3 ANKRD17 EPHA7 KIAA0746 BMP3 BRDG1 GABRR1 ANAPC4 BTC SMR3A GABRR2 SLA/LP CCNG2 ASAHL GRIK2 LGI2 SCARB2 COQ2 HTR1B TBC1D19 CDS1 SULT1B1 HTR1E PI4K2B CENPC1 TMPRSS11E IMPG1 STIM2 CSN1S1 MRPS18C ME1 GBA3 CSN2 COPS4 NT5E KCNIP4 CSN3 HSD17B11 PGM3 DKFZp761B107 DCK HERC5 POU3F2 C4orf28 DMP1 PLAC8 PREP GPR125 DSPP NUDT9 SIM1 FLJ45721 EPHA5 NUP54 ELOVL4 hsa-mir-218-1 EREG UGT2B28 MAP3K7 FGF5 ODAM TPBG GC HERC6 TTK GK2 SDAD1 RNGTT GNRHR UBE1L2 TBX18 -

An Evolutionary Medicine Perspective on Neandertal Extinction
Supplementary Information (Figures and Tables) for: An evolutionary medicine perspective on Neandertal extinction Alexis P. Sullivan1, Marc de Manuel3, Tomas Marques-Bonet3,4,5, & George H. Perry1,2 Departments of 1Biology and 2Anthropology, Pennsylvania State University, University Park, PA 16802, USA 3Institut de Biologia Evolutiva (CSIC/UPF), Parque de Investigación Biomédica de Barcelona (PRBB), Barcelona, Catalonia 08003, Spain 4CNAG-CRG, Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology (BIST), Baldiri i Reixac 4, 08028 Barcelona, Spain 5Catalan Institution of Research and Advanced Studies (ICREA), Passeig de Lluís Companys, 23, 08010, Barcelona, Spain Corresponding Author: George H. Perry E-mail: [email protected] Supplemental Figure 1: Innate immune system gene permutation analyses – 10,000 sets of 73 randomly selected genes containing nonsynonymous SNPs Supplemental Figure 2: Virus-interacting protein gene permutation analyses – 10,000 sets of 164 randomly selected genes containing nonsynonymous SNPs Supplemental Figure 3: MHC gene permutation analyses – 10,000 sets of 13 randomly selected genes containing nonsynonymous SNPs Supplemental Figure 4: Patterns of Neandertal and modern human nonsynonymous SNP diversity in MHC genes (n = 13) excluding the Altai Neandertal and one random modern human per population Supplemental Figure 5: Significantly enriched gene ontology categories (red) among top 1% ape diversity genes Supplemental Table 1: A comparison of genome-wide nonsynonymous SNPs versus total (nonsynonymous -

1 CCR-11-2535 Re-Revised Title: Integrated Analysis Reveals Critical
Author Manuscript Published OnlineFirst on January 24, 2012; DOI: 10.1158/1078-0432.CCR-11-2535 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. CCR-11-2535 Re-Revised Title: Integrated Analysis Reveals Critical Genomic Regions in Prostate Tumor- Microenvironment Associated with Clinico-pathologic Phenotypes Authors: Shingo Ashida1, Mohammed S. Orloff1,3, Gurkan Bebek1,8,10, Li Zhang1,2, Pan Zheng5,6, Donna M. Peehl7 and Charis Eng1,3,4,9,10 Affiliations: 1Genomic Medicine Institute and 2Section of Statistical Genetics and Bioinformatics, Department of Quantitative Health Sciences, Lerner Research Institute, 3Taussig Cancer Institute, and 4Stanley Shalom Zielony Institute for Nursing Excellence, Cleveland Clinic, Cleveland, Ohio; 5Department of Pathology, The Ohio State University, Columbus, Ohio; 6Division of Immunotherapy, Department of Surgery, and Program of Molecular Medicine, Comprehensive Cancer Center, University of Michigan Medical Center, Ann Arbor, Michigan; 7Department of Urology, Stanford University School of Medicine, Stanford, California; 8Center for Proteomics and Bioinformatics, 9Department of Genetics and 10CASE Comprehensive Cancer Center, Case Western Reserve University School of Medicine, Cleveland, Ohio Corresponding Author: Charis Eng, MD, PhD, Cleveland Clinic Genomic Medicine Institute, 9500 Euclid Avenue, Mailstop NE-50, Cleveland, OH 44195 (email: [email protected]) Running Title: Integrated genome-wide analyses for prostate cancer-stroma Key Words: Prostate tumor-stroma, LOH, clinico-pathologic correlates 1 Downloaded from clincancerres.aacrjournals.org on September 30, 2021. © 2012 American Association for Cancer Research. Author Manuscript Published OnlineFirst on January 24, 2012; DOI: 10.1158/1078-0432.CCR-11-2535 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

Supplementary Materialsupplementary Material
DOI:10.1071/AN20275_AC CSIRO 2021 Animal Production Science 2021, 61, 731–744 Genome-wide association study between copy number variation regions and carcass and meat quality traits in Nellore cattle Mariana Piatto Berton1,5, Marcos Vinícius de Antunes Lemos1, Tatiane Cristina Seleguim Chud2, Nedenia Bonvino Stafuzza1, Sabrina Kluska1, Sabrina Thaise Amorim1, Lucas Silva Ferlin Lopes1, Angélica Simone Cravo Pereira3, Derek Bickhart4, George Liu4, Lúcia Galvão de Albuquerque1 and Fernando Baldi1 1Departamento de Zootecnia, Faculdade de Ciências Agrárias e Veterinárias, Universidade Estadual Paulista, Via de acesso Prof. Paulo Donato Castellane, s/no, CEP 14884-900 Jaboticabal, SP, Brazil 2Centre for Genetic Improvement of Livestock, University of Guelph, Guelph, ON, Canada N1G 2W1 3 Faculdade de Zootecnia e Engenharia de Alimentos, Universidade de São Paulo, Rua Duque de Caxias Norte, 225, CEP 13635-900 Pirassununga, SP, Brazil 4 USDA-ARS, ANRI, Bovine Functional Genomics Laboratory, Beltsville, Maryland 20705, USA; 2Department of Animal and Avian Sciences, University of Maryland, College Park, Maryland 20742, USA 5Corresponding author. E-mail: [email protected] Table S1. Description of significant (P value<0.05) CNVRs associated with beef tenderness (BT) and candidate genes within the CNVRs. CNVR ID BTA Type* Start (bp) End (bp) Size (bp) Low** High** P value FDR CNVR_gain_3 1 Gain 1374155 3404571 2030417 27 45 0.026 0.36 CNVR_gain_644 2 Gain 81291862 83154452 1862591 32 15 0.015 0.40 CNVR_loss_936 5 Loss 22514133 22563988 49856 233 -

Supplemental Information
Supplemental information Supplemental Material and Methods Isolation of mononuclear cells from the frest ovarian tumor specimens Fresh ovarian tumor specimens were minced with scissors, digested in PBS containing 1 mg/mL of Collagenase D (Roche) and 100μg/mL DNase I at 37°C for 30 min, mechanically dissociated using the gentleMACS dissociator (Miltenyi Biotec) and passed through a 100µm nylon cell strainer (BD Biosciences). Degranulation and IFNγ production after in vitro stimulation Mononuclear cells isolated from fresh tumor specimens were stimulated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA) + 1 μg/mL ionomycin in the presence of anti-CD107a FITC monoclonal antibody (BioLegend) for 1h followed by 3h incubation with brefeldin A (BioLegend). Unstimulated cells were used as control. Cells were then washed in PBS, stained with anti-CD45 PerCP (EXBIO), anti-CD3 Alexa Fluor 700 (EXBIO), anti-CD4 ECD (Beckman Coulter) and anti-CD8 HV500 (BD Biosciences) monoclonal antibodies, fixed in fixation/permeabilization buffer (eBioscience), further permeabilized with permeabilization buffer (eBioscience) and stained with anti-IFNγ PE-Cy7 (eBioscience), anti-GZMB Brilliant Violet 421 (BD Biosciences) and anti-PRF1 APC (BioLegend) monoclonal antibodies. The percentage of CD3+CD8+ T cells producing IFNγ and degranulating upon PMA/ionomycin stimulation were determined by flow cytometry. The data were analyzed with the FlowJo software package (Tree Star, Inc.). “Expression profile of co-inhibitory receptors on CD8+ T cells upon exposure to rIFNγ in vitro” Mononuclear and malignant cells were isolated from fresh HGSC specimens and cultured with rIFNγ under different conditions (Supplementary Fig. 6A). (1) Malignant cells and leukocytes were co-cultured in presence or absence of 20ng/mL rIFNγ for 24 hours. -
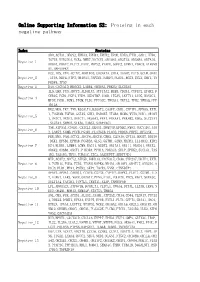
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

Supp Info First Page
Supplementary Information (Figures and Tables) for: An evolutionary medicine perspective on Neandertal extinction Alexis P. Sullivan1, Marc de Manuel3, Tomas Marques-Bonet3,4,5, & George H. Perry1,2 Departments of 1Biology and 2Anthropology, Pennsylvania State University, University Park, PA 16802, USA 3Institut de Biologia Evolutiva (CSIC/UPF), Parque de Investigación Biomédica de Barcelona (PRBB), Barcelona, Catalonia 08003, Spain 4CNAG-CRG, Centre for Genomic Regulation (CRG), Barcelona Institute of Science and Technology (BIST), Baldiri i Reixac 4, 08028 Barcelona, Spain 5Catalan Institution of Research and Advanced Studies (ICREA), Passeig de Lluís Companys, 23, 08010, Barcelona, Spain Corresponding Author: George H. Perry E-mail: [email protected] Supplemental Figure 1: Innate immune system gene permutation analyses – 10,000 sets of 73 randomly selected genes containing nonsynonymous SNPs Supplemental Figure 2: MHC gene permutation analyses – 10,000 sets of 13 randomly selected genes containing nonsynonymous SNPs Supplemental Figure 3: Significantly enriched gene ontology categories (red) among top 1% ape diversity genes Supplemental Table 1: A comparison of genome-wide nonsynonymous SNPs versus total (nonsynonymous + synonymous) SNPs between Neandertal and modern human populations Supplemental Table 2: PolyPhen-2 predictions for genome-wide nonsynonymous SNPs – damaging versus not damaging – for Neandertal and modern human populations Supplemental Table 3: List of Innate immune system genes Supplemental Table 4: List of MHC genes Supplemental -

Supplementary Methods
doi: 10.1038/nature06162 SUPPLEMENTARY INFORMATION Supplementary Methods Cloning of human odorant receptors 423 human odorant receptors were cloned with sequence information from The Olfactory Receptor Database (http://senselab.med.yale.edu/senselab/ORDB/default.asp). Of these, 335 were predicted to encode functional receptors, 45 were predicted to encode pseudogenes, 29 were putative variant pairs of the same genes, and 14 were duplicates. We adopted the nomenclature proposed by Doron Lancet 1. OR7D4 and the six intact odorant receptor genes in the OR7D4 gene cluster (OR1M1, OR7G2, OR7G1, OR7G3, OR7D2, and OR7E24) were used for functional analyses. SNPs in these odorant receptors were identified from the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP) or through genotyping. OR7D4 single nucleotide variants were generated by cloning the reference sequence from a subject or by inducing polymorphic SNPs by site-directed mutagenesis using overlap extension PCR. Single nucleotide and frameshift variants for the six intact odorant receptors in the same gene cluster as OR7D4 were generated by cloning the respective genes from the genomic DNA of each subject. The chimpanzee OR7D4 orthologue was amplified from chimpanzee genomic DNA (Coriell Cell Repositories). Odorant receptors that contain the first 20 amino acids of human rhodopsin tag 2 in pCI (Promega) were expressed in the Hana3A cell line along with a short form of mRTP1 called RTP1S, (M37 to the C-terminal end), which enhances functional expression of the odorant receptors 3. For experiments with untagged odorant receptors, OR7D4 RT and S84N variants without the Rho tag were cloned into pCI.