Article Enigmatic Orthology Relationships Between Hox
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

13914444D46c0aa91d02e31218
2 Breeding of wild and some domestic animals at regional zoological institutions in 2013 3 РЫБЫ P I S C E S ВОББЕЛОНГООБРАЗНЫЕ ORECTOLOBIFORMES Сем. Азиатские кошачьи акулы (Бамбуковые акулы) – Hemiscyllidae Коричневополосая бамбуковая акула – Chiloscyllium punctatum Brownbanded bambooshark IUCN (NT) Sevastopol 20 ХВОСТОКОЛООБРАЗНЫЕ DASYATIFORMES Сем. Речные хвостоколы – Potamotrygonidae Глазчатый хвостокол (Моторо) – Potamotrygon motoro IUCN (DD) Ocellate river stingray Sevastopol - ? КАРПООБРАЗНЫЕ CYPRINIFORMES Сем. Цитариновые – Citharinidae Серебристый дистиход – Distichodusaffinis (noboli) Silver distichodus Novosibirsk 40 Сем. Пираньевые – Serrasalmidae Серебристый метиннис – Metynnis argenteus Silver dollar Yaroslavl 10 Обыкновенный метиннис – Metynnis schreitmuelleri (hypsauchen) Plainsilver dollar Nikolaev 4; Novosibirsk 100; Kharkov 20 Пятнистый метиннис – Metynnis maculatus Spotted metynnis Novosibirsk 50 Пиранья Наттерера – Serrasalmus nattereri Red piranha Novosibirsk 80; Kharkov 30 4 Сем. Харацидовые – Characidae Красноплавничный афиохаракс – Aphyocharax anisitsi (rubripinnis) Bloodfin tetra Киев 5; Perm 10 Парагвайский афиохаракс – Aphyocharax paraquayensis Whitespot tetra Perm 11 Рубиновый афиохаракс Рэтбина – Aphyocharax rathbuni Redflank bloodfin Perm 10 Эквадорская тетра – Astyanax sp. Tetra Perm 17 Слепая рыбка – Astyanax fasciatus mexicanus (Anoptichthys jordani) Mexican tetra Kharkov 10 Рублик-монетка – Ctenobrycon spilurus (+ С. spilurusvar. albino) Silver tetra Kharkov 20 Тернеция (Траурная тетра) – Gymnocorymbus -

Whole Genome Sequencing of the Asian Arowana (Scleropages Formosus) Provides Insights Into the Evolution of Ray-Finned Fishes
GBE Whole Genome Sequencing of the Asian Arowana (Scleropages formosus) Provides Insights into the Evolution of Ray-Finned Fishes Christopher M. Austin1,2,y, Mun Hua Tan1,2,y, Larry J. Croft1,2,3, Michael P. Hammer4,and HanMingGan1,2,* 1School of Science, Monash University Malaysia, Petaling Jaya, Selangor, Malaysia 2Monash University Malaysia Genomics Facility, Monash University Malaysia, Petaling Jaya, Selangor, Malaysia 3Malaysian Genomics Resource Centre Berhad, Boulevard Signature Office, Kuala Lumpur, Malaysia 4Museum and Art Gallery of the Northern Territory, Darwin, NT, Australia *Corresponding author: E-mail: [email protected]. yThese authors contributed equally to this work. Accepted: September 28, 2015 Data deposition: This project has been deposited at DNA Data Bank of Japan/EMBL/GenBank under the accession JARO00000000. Abstract The Asian arowana (Scleropages formosus) is of commercial importance, conservation concern, and is a representative of one of the oldest lineages of ray-finned fish, the Osteoglossomorpha. To add to genomic knowledge of this species and the evolution of teleosts, the genome of a Malaysian specimen of arowana was sequenced. A draft genome is presented consisting of 42,110 scaffolds with a total size of 708 Mb (2.85% gaps) representing 93.95% of core eukaryotic genes. Using a k-mer-based method, a genome size of 900 Mb was also estimated. We present an update on the phylogenomics of fishes based on a total of 27 species (23 fish species and 4 tetrapods) using 177 orthologous proteins (71,360 amino acid sites), which supports established relationships except that arowana is placed as the sister lineage to all teleost clades (Bayesian posterior probability 1.00, bootstrap replicate 93%), that evolved after the teleost genome duplication event rather than the eels (Elopomorpha). -
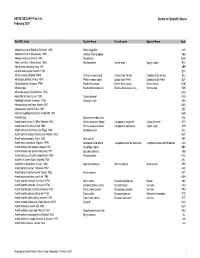
ASFIS ISSCAAP Fish List February 2007 Sorted on Scientific Name
ASFIS ISSCAAP Fish List Sorted on Scientific Name February 2007 Scientific name English Name French name Spanish Name Code Abalistes stellaris (Bloch & Schneider 1801) Starry triggerfish AJS Abbottina rivularis (Basilewsky 1855) Chinese false gudgeon ABB Ablabys binotatus (Peters 1855) Redskinfish ABW Ablennes hians (Valenciennes 1846) Flat needlefish Orphie plate Agujón sable BAF Aborichthys elongatus Hora 1921 ABE Abralia andamanika Goodrich 1898 BLK Abralia veranyi (Rüppell 1844) Verany's enope squid Encornet de Verany Enoploluria de Verany BLJ Abraliopsis pfefferi (Verany 1837) Pfeffer's enope squid Encornet de Pfeffer Enoploluria de Pfeffer BJF Abramis brama (Linnaeus 1758) Freshwater bream Brème d'eau douce Brema común FBM Abramis spp Freshwater breams nei Brèmes d'eau douce nca Bremas nep FBR Abramites eques (Steindachner 1878) ABQ Abudefduf luridus (Cuvier 1830) Canary damsel AUU Abudefduf saxatilis (Linnaeus 1758) Sergeant-major ABU Abyssobrotula galatheae Nielsen 1977 OAG Abyssocottus elochini Taliev 1955 AEZ Abythites lepidogenys (Smith & Radcliffe 1913) AHD Acanella spp Branched bamboo coral KQL Acanthacaris caeca (A. Milne Edwards 1881) Atlantic deep-sea lobster Langoustine arganelle Cigala de fondo NTK Acanthacaris tenuimana Bate 1888 Prickly deep-sea lobster Langoustine spinuleuse Cigala raspa NHI Acanthalburnus microlepis (De Filippi 1861) Blackbrow bleak AHL Acanthaphritis barbata (Okamura & Kishida 1963) NHT Acantharchus pomotis (Baird 1855) Mud sunfish AKP Acanthaxius caespitosa (Squires 1979) Deepwater mud lobster Langouste -

Informational Issue of Eurasian Regional Association of Zoos and Aquariums
GOVERNMENT OF MOSCOW DEPARTMENT FOR CULTURE EURASIAN REGIONAL ASSOCIATION OF ZOOS & AQUARIUMS MOSCOW ZOO INFORMATIONAL ISSUE OF EURASIAN REGIONAL ASSOCIATION OF ZOOS AND AQUARIUMS VOLUME № 28 MOSCOW 2009 GOVERNMENT OF MOSCOW DEPARTMENT FOR CULTURE EURASIAN REGIONAL ASSOCIATION OF ZOOS & AQUARIUMS MOSCOW ZOO INFORMATIONAL ISSUE OF EURASIAN REGIONAL ASSOCIATION OF ZOOS AND AQUARIUMS VOLUME № 28 _________________ MOSCOW - 2009 - Information Issue of Eurasian Regional Association of Zoos and Aquariums. Issue 28. – 2009. - 424 p. ISBN 978-5-904012-10-6 The current issue comprises information on EARAZA member zoos and other zoological institutions. The first part of the publication includes collection inventories and data on breeding in all zoological collections. The second part of the issue contains information on the meetings, workshops, trips and conferences which were held both in our country and abroad, as well as reports on the EARAZA activities. Chief executive editor Vladimir Spitsin General Director of Moscow Zoo Compiling Editors: Т. Andreeva M. Goretskaya N. Karpov V. Ostapenko V. Sheveleva T. Vershinina Translators: T. Arzhanova M. Proutkina A. Simonova УДК [597.6/599:639.1.04]:59.006 ISBN 978-5-904012-10-6 © 2009 Moscow Zoo Eurasian Regional Association of Zoos and Aquariums Dear Colleagues, (EARAZA) We offer you the 28th volume of the “Informational Issue of the Eurasian Regional Association of Zoos and Aquariums”. It has been prepared by the EARAZA Zoo 123242 Russia, Moscow, Bolshaya Gruzinskaya 1. Informational Center (ZIC), based on the results of the analysis of the data provided by Telephone/fax: (499) 255-63-64 the zoological institutions of the region. E-mail: [email protected], [email protected], [email protected]. -

Aspects of the Biology and Culture of the Butterfly Fish, Pantodon Buchholzi; a Potential Aquarium Fish in Nigeria
Full-text Available Online at J. Appl. Sci. Environ. Manage. PRINT ISSN 1119-8362 Electronic ISSN 1119-8362 https://www.ajol.info/index.php/jasem Vol. 23 (7) 1273-1277 July 2019 http://ww.bioline.org.br/ja Aspects of the Biology and Culture of the Butterfly Fish, Pantodon buchholzi; A Potential Aquarium Fish in Nigeria *IBIM, AT; IKE, JO Department of Fisheries, Faculty of Agriculture, University of Port Harcourt, Choba, Nigeria *Corresponding Author Email: [email protected] ABSTRACT: A review was carried out on a Nigerian fish species with ornamental importance, the Pantodon buchholzi. This was aimed at introducing it an additional aquarium fish and eliciting interest in the large scale culture of the species in Nigeria, to develop the Ornamental Fishery Industry. The study on the biology revealed that the species were spectacular species, possessing unique physical features that confer on them the ornamental status. They are widely distributed in flooded vegetated acidic freshwater environment in Nigeria and some other West African Countries. They require optimum water conditions of 25-27oC, pH of 6.5 -7.0, and hardness of 8-12. The adults are carnivorous, surface hunters, while the juveniles are passive insectivores that prefer live foods, but can be lured into eating fish feed over time. They mature sexually in a year or more. Information on the culture is scarce except for experiences of a few Aquarium fish keepers or hobbyists, who reported that they are hardy, but do not spawn readily in captivity. Also, they are community fishes but can be cannibalistic towards other fishes, and their young too. -

One Flexible Source Suitable for Many Different Readers World Book Is A
One flexible source suitable for many different readers World Book is a reliable source of factual information, a basic for school assignments and everyday reference needs. But it also can prompt curiosity and widen imagination; it can excite readers of all ages and lead them to study and learn more. To effectively fulfill these roles for readers’ varied comprehension skills and varying information needs, World Book is designed to be a flexible source. Librarians and teachers know best their readers’ and students’ interests, needs, and skills. They perceive when a reader is ready to stretch or when a student needs support. World Book’s flexibility enables librarians and teachers to respond to a range of reader and student uses with a single convenient and trusted source. Article structure and features The “Fish” article is an example of how World Book crafts content to be appropriate for many readers’ needs. The following is characteristic of all World Book’s longer articles: • The introductory paragraphs highlight the most important aspects of the topic. The introduction to this article tells what fish are and where they live; their importance to humans, common physical features, and natural history. This may be all a reader wants or needs to know. • The outline is available to alert the reader to the remainder of the article and guide the reader through it. • World Book organizes many major articles into sections called “topical units.” Each topical unit stands on its own within the context of the larger article, following the principle that, for effective learning, subjects should be broken down into manageable units—particularly useful when a reader has a limited amount of time or a limited attention span. -

Regional Biosecurity Plan for Micronesia and Hawaii Volume II
Regional Biosecurity Plan for Micronesia and Hawaii Volume II Prepared by: University of Guam and the Secretariat of the Pacific Community 2014 This plan was prepared in conjunction with representatives from various countries at various levels including federal/national, state/territory/commonwealth, industry, and non-governmental organizations and was generously funded and supported by the Commander, Navy Installations Command (CNIC) and Headquarters, Marine Corps. MBP PHASE 1 EXECUTIVE SUMMARY NISC Executive Summary Prepared by the National Invasive Species Council On March 7th, 2007 the U.S. Department of Navy (DoN) issued a Notice of Intent to prepare an “Environmental Impact Statement (EIS)/Overseas Environmental Impact Statement (OEIS)” for the “Relocation of U.S. Marine Corps Forces to Guam, Enhancement of Infrastructure and Logistic Capabilities, Improvement of Pier/Waterfront Infrastructure for Transient U.S. Navy Nuclear Aircraft Carrier (CVN) at Naval Base Guam, and Placement of a U.S. Army Ballistic Missile Defense (BMD) Task Force in Guam”. This relocation effort has become known as the “build-up”. In considering some of the environmental consequences of such an undertaking, it quickly became apparent that one of the primary regional concerns of such a move was the potential for unintentional movement of invasive species to new locations in the region. Guam has already suffered the eradication of many of its native species due to the introduction of brown treesnakes and many other invasive plants, animals and pathogens cause tremendous damage to its economy and marine, freshwater and terrestrial ecosystems. DoN, in consultation and concurrence with relevant federal and territorial regulatory entities, determined that there was a need to develop a biosecurity plan to address these concerns. -

EAZA Freshwater Teleost TAG Regional Collection Plan
EAZA Freshwater teleost TAG Regional Collection Plan December 2020 1st Edition Editors Anton Weissenbacher | Tiergarten Schönbrunn, chair EAZA Freshwater teleost TAG Brian Zimmerman | ZSL London Zoo, Advisor EAZA Freshwater teleost TAG David Aparici Plaza | EAZA Executive Office, TAG liaison Elmar Fienieg| EAZA Executive Office, Population biologist Nora Hausen| EAZA Executive Office, Assistant population biologist Citation Weissenbacher, A., Zimmerman, B., Aparici Plaza, D., Fienieg, E., Hausen, N. (eds.) 2020. Regional Collection Plan –EAZA Freshwater Teleost Taxon Advisory Group– Edition One. EAZA Executive Office: Amsterdam. Cover photo credit: European Mudminnow (Umbra krameri) by Barbara Nicca, Jordan mouthbrooder (Astatotilapia flaviijosephi) by Barbara Nicca, and Valencia robertae (Valencia robertae) by Yannis Kapakos. Acknowledgements This final report of the Regional Collection Plan for the EAZA Freshwater Teleost Taxon Advisory Group is the result of a collaboration of many people involved. This is a product of the EAZA Freshwater Teleost TAG, with input and support from the workshop participants and staff of the EAZA Executive Office. The EAZA Freshwater Teleost RCP workshop took place on 12 to 14 November 2019 in EAZA Executive Office, Amsterdam (The Netherlands) and was organised by the EAZA Freshwater Teleost TAG and the EAZA Executive Office. Freshwater teleost TAG mission statement: “To achieve conservation by managing freshwater teleost populations that mainly function as Ark or Rescue populations. A large number of freshwater fishes are threatened with extinction and several are already extinct in the wild. For many of these species ex situ populations can be maintained with relatively few resources and high chances of a successful reintroduction to the wild in the future. -

Whole Genome Sequencing of the Pirarucu (Arapaima Gigas) Supports Independent Emergence of Major Teleost Clades
1/36 Whole genome sequencing of the Pirarucu (Arapaima gigas) supports independent emergence of major teleost clades Authors: Ricardo Assunção Vialle1†, Jorge Estefano Santana de Souza2†, Katia de Paiva Lopes1, Diego Gomes Teixeira2, Pitágoras de Azevedo Alves Sobrinho2, André M. Ribeiro-dos-Santos1,3,Carolina Furtado4, Tetsu Sakamoto5, Fábio Augusto Oliveira Silva6, Edivaldo Herculano Corrêa de Oliveira6, Igor Guerreiro Hamoy7, Paulo Pimentel Assumpção8, Ândrea Ribeiro-dos-Santos1,8, João Paulo Matos Santos Lima2,9, Héctor N. Seuánez4,10,Sandro José de Souza2,11, Sidney Santos1,8* Affiliations: 1Laboratório de Genética Humana e Médica, Instituto de Ciências Biológicas, Universidade Federal do Pará, Belém, PA, Brazil 2Bioinformatics Multidisciplinary Environment – BioME, Universidade Federal do Rio Grande do Norte, Natal, RN, Brazil 3Departmento de Genética, Universidade Federal do Rio Grande do Norte, Natal, RN, Brazil 4Programa de Genética, Instituto Nacional de Câncer (INCA), Rio de Janeiro, RJ, Brazil 5Instituto de Ciências Biológicas, Departamento de Bioquímica e Imunologia, Universidade Federal de Minas Gerais, Belo Horizonte, MG, Brazil 6Laboratório de Cultura de Tecidos e Citogenética, Instituto Evandro Chagas, Belém, PA, Brazil 7Laboratório de Genética Aplicada, Universidade Federal Rural da Amazônia, Belém, PA, Brazil 8Núcleo de Pesquisas em Oncologia, Universidade Federal do Pará, Belém, PA, Brazil 9Departamento de Bioquímica, Universidade Federal do Rio Grande do Norte, Natal, RN, Brazil 10Departamento de Genética, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, Brazil 11Instituto do Cérebro, Universidade Federal do Rio Grande do Norte, Natal, RN, Brazil * Author for correspondence: Sidney Santos1,8* - [email protected] † These authors contributed equally to this work. Data deposition: This project has been deposited at EBI-ENA under accession PRJEB22808. -

Possible Multiple Evolution of Indirect Telencephalo-Cerebellar Pathways in Teleosts: Studies in Carassius Auratus and Pantodon Buchholzi
Cell Tissue Res (1993) 274:447-455 Cell Tissue Research Springer-Verlag 1993 Possible multiple evolution of indirect telencephalo-cerebellar pathways in teleosts: studies in Carassius auratus and Pantodon buchholzi Mario F. Wullimann 1, Dietrich L. Meyer 2 i Institut fiir Hirnforschung,Universit/it Bremen, FB 2-NW II, Postfach 33 04 40, D-28334 Bremen, Germany 2 Abteilung Neuroanatomie, Zentrum Anatomie, Universit/it G6ttingen, D-37075 GSttingen, Germany Received: 1 March 1993 / Accepted: 30 April 1993 Abstract. Among vertebrates, telencephalo-pontine sys- pendently (Roth and Wullimann, in press). In contrast to tems exist only in birds and mammals. However, three birds and mammals, teleost fishes lack a pons. However, nuclei in the diencephalon and mesencephalon of teleost three nuclei in the diencephalon and mesencephalon of fishes have been indicated analogous to the pons to various teleost species (see Discussion) have been indicat- represent relay stations between telencephalon and cere- ed as relay stations for telencephalo-cerebellar pathways. bellum. Since two of these nuclei (dorsal preglomerular (1) Some euteleosts, the evolutionarily most derived nucleus, dorsal tegmental nucleus) have only been de- teleost clade (Fig. 5), possess a telencephalo-cerebellar scribed in the highly derived, electrosensory mormyrids, pathway via the nucleus paracommissuralis, which is lo- we investigated telencephalic connections in two non- cated most dorsally in the diencephalon (Karten and Fin- electrosensory teleosts, the goldfish Carassius auratus ger 1976; Ito et al. 1982; Striedter 1990). The nucleus and and the freshwater butterflyfish Pantodon buchholzi, and the related pathway have only been described in eu- cerebellar connections only in the latter species, since for teleosts (Wullimann and Northcutt 1988). -

Paratilapia Polleni Pollen’S Cichlid 3Rd Place Winner Judith Weinberg’S Entry in the 39Th NEC Convention Fish Show 1 103 Y EARS of E DUCATING a QUARISTS AQUATICA VOL
QUATICAQU AT H E O N - L I N E J O U R N A L O F T H E B R O O K L Y N A Q U A R I U M S O C I E T Y VOL. 28 SEPTEMBER ~ OCTOBER 2014 No. 1 Paratilapia polleni Pollen’s cichlid 3rd place winner Judith Weinberg’s entry in the 39th NEC Convention fish show 1 103 Y EARS OF E DUCATING A QUARISTS AQUATICA VOL. 28 SEPTEMBER - OCTOBER 2014 NO. 1 C ONTENT S PAGE 2 THE AQUATICA STAFF what they need in your reef tank. GERALD JENNINGS & JERRY HALL PAGE 3 CALENDAR OF EVENTS BAS Events for the years 2014 - 2015. PAGE 21 AN EXPERIENCE WITH WHIPTAIL CATFISH OF THE RINELORICARIA. PAGE 4 BAS 2014 PHOTO CONTEST. GENUS Ian’s The rules for our second photo contest. experience with breeding this interesting Take your best shot and send it in...you species of catfish. may be a winner. IAN FULLER, BAS BAS Board PAGE 24 SPECIES PROFILE. The PAGE 5 ONE-OF-A-KIND. Learn Whiptail catfish, Rineloricaria hasemani. more about the Butterflyfish which is JOHN TODARO, BAS the only species in the genus Pantodon. PAGE 25 CATFISH CONNECTIONS. JOHN TODARO, BAS Introducing a new column for you catfish enthusiasts. PAGE 7 SPECIES PROFILE. The Butterflyfish, SY ANGELICUS, BAS Pantodon buchholzi . JOHN TODARO, BAS PAGE 26 SPECIES PROFILE. Angelicus catfish, Synodontis angelicus. PAGE 8 WHAT IS THE BEST WAY TO JOHN TODARO, BAS “CYCLE” A REEF TANK? Directions on how to cycle a reef tank. -

The Ichthyological Diversity of Pokémon
The ichthyological diversity of Pokémon Augusto B. Mendes1, Felipe V. Guimarães2, Clara B. P. Eirado-Silva1 & Edson P. Silva1 1Universidade Federal Fluminense, Niterói, RJ, Brazil. 2Universidade do Estado do Rio de Janeiro, São Gonçalo, RJ, Brazil. Emails: [email protected]; [email protected]; [email protected]; [email protected] Pokémon, or Pocket Monsters, was 1998, selling together more than 10 million originally created for videogames, becoming a copies. Also in 1998, the Yellow version of the worldwide fever among kids and teenagers in game was released, which has as its most the end of the 1990’s and early 2000’s. distinct feature the possibility of having Pikachu Currently, it is still a success, with numerous (the most famous Pokémon) walking side by games, a TV series, comic books, movies, a side with the player in the game. Pokémon Trading Card Game, toys and collectibles. Green, Red, Blue and Yellow are the so-called Through its core products and vibrant “first generation” of games in the franchise. merchandising, Pokémon took over the world, Today, the Pokémon series is in its seventh influencing pop culture wherever it landed. generation, with 29 main games released, Despite losing some steam in the early 2010’s, besides several spin-offs. The TV series, on the Pokémon is now back to its previous uproar other hand, is in its sixth season, with more with the release of Pokémon GO, an augmented than 900 episodes. reality (AR) game for smartphones. This game The games and TV series take place in launched in 2016, with almost 21 million users regions inhabited by many Pokémon and downloading it in the very first week in the humans.