Altered RNA Processing in Cancer Pathogenesis and Therapy
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

SF3B3) and Sin3a Associated Protein 130 (SAP130
cells Communication Ambiguity about Splicing Factor 3b Subunit 3 (SF3B3) and Sin3A Associated Protein 130 (SAP130) Paula I. Metselaar 1,* , Celine Hos 1, Olaf Welting 1, Jos A. Bosch 2,3, Aletta D. Kraneveld 4 , Wouter J. de Jonge 1 and Anje A. Te Velde 1 1 Tytgat Institute for Liver and Intestinal Research, AGEM, Amsterdam UMC, University of Amsterdam, 1105BK Amsterdam, The Netherlands; [email protected] (C.H.); [email protected] (O.W.); [email protected] (W.J.d.J.); [email protected] (A.A.T.V.) 2 Department of Psychology, University of Amsterdam, 1018WS Amsterdam, The Netherlands; [email protected] 3 Department of Medical Psychology, Amsterdam UMC, University of Amsterdam, 1001NK Amsterdam, The Netherlands 4 Division of Pharmacology, Utrecht Institute for Pharmaceutical Sciences, Faculty of Science, Utrecht University, 3584CG Utrecht, The Netherlands; [email protected] * Correspondence: [email protected] Abstract: In 2020, three articles were published on a protein that can activate the immune system by binding to macrophage-inducible C-type lectin receptor (Mincle). In the articles, the protein was referred to as ‘SAP130, a subunit of the histone deacetylase complex.’ However, the Mincle ligand the authors aimed to investigate is splicing factor 3b subunit 3 (SF3B3). This splicing factor is unrelated to SAP130 (Sin3A associated protein 130, a subunit of the histone deacetylase-dependent Sin3A corepressor complex). The conclusions in the three articles were formulated for SF3B3, Citation: Metselaar, P.I.; Hos, C.; while the researchers used qPCR primers and antibodies against SAP130. -

Family Member in Teleost Fish Identification of a Novel IL-1 Cytokine
The Journal of Immunology Identification of a Novel IL-1 Cytokine Family Member in Teleost Fish1 Tiehui Wang,* Steve Bird,* Antonis Koussounadis,† Jason W. Holland,* Allison Carrington,* Jun Zou,* and Christopher J. Secombes2* A novel IL-1 family member (nIL-1F) has been discovered in fish, adding a further member to this cytokine family. The unique gene organization of nIL-1F, together with its location in the genome and low homology to known family members, suggests that this molecule is not homologous to known IL-1F. Nevertheless, it contains a predicted C-terminal -trefoil structure, an IL-1F signature region within the final exon, a potential IL-1 converting enzyme cut site, and its expression level is clearly increased following infection, or stimulation of macrophages with LPS or IL-1. A thrombin cut site is also present and may have functional relevance. The C-terminal recombinant protein antagonized the effects of rainbow trout rIL-1 on inflammatory gene expression in a trout macrophage cell line, suggesting it is an IL-1 antagonist. Modeling studies confirmed that nIL-1F has the potential to bind to the trout IL-1RI receptor protein, and may be a novel IL-1 receptor antagonist. The Journal of Immunology, 2009, 183: 962–974. he IL-1 family (IL-1F)3 of cytokines is characterized by of transcription factors such as NF-B and MAPK-regulated tran- their common secondary structure of an all- fold, the scription factors, leading to IL-1F-regulated gene transcription in T -trefoil, which they have in common with another cyto- the target cells (1). -

The Dawn of Next Generation DNA Sequencing in Myelodysplastic Syndromes- Experience from Pakistan
The Dawn Of Next Generation DNA Sequencing In Myelodysplastic Syndromes- Experience From Pakistan. Nida Anwar ( [email protected] ) National Institute of Blood Diseases and Bone Marrow Transplantation Faheem Ahmed Memon Liaquat University of Medical & Health Sciences Saba Shahid National Institute of Blood Diseases and Bone Marrow Transplantation Muhammad Shakeel Jamil-ur-Rahman Center for Genome Research, University of Karachi Muhammad Irfan Jamil-ur-Rahman Center for Genome Research, University of Karachi Aisha Arshad National Institute of Blood Diseases and Bone Marrow Transplantation Arshi Naz Liaquat University of Medical & Health Sciences Ikram Din Ujjan Liaquat University of Medical & Health Sciences Tahir Shamsi National Institute of Blood Diseases and Bone Marrow Transplantation Research Article Keywords: Myelodysplastic Syndromes, Next generation sequencing, Gene analysis, Mutation, Pakistan. Posted Date: June 8th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-571430/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/16 Abstract Background: Myelodysplastic syndromes (MDS) are clonal disorders of hematopoietic stem cells exhibiting ineffective hematopoiesis and tendency for transformation into acute myeloid leukemia (AML). The available karyotyping and uorescent in situ hybridization provide limited information on molecular abnormalities for diagnosis/prognosis of MDS. Next generation DNA sequencing (NGS), providing deep insights into molecular mechanisms being involved in pathophysiology, was employed to study MDS in Pakistani cohort. Patients and Methods: It was a descriptive cross-sectional study carried out at National institute of blood diseases and bone marrow transplant from 2016 to 2019. Total of 22 cases of MDS were included. Complete blood counts, bone marrow assessment and cytogenetic analysis was done. -

SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression
Published OnlineFirst August 20, 2019; DOI: 10.1158/0008-5472.CAN-18-3965 Cancer Molecular Cell Biology Research SF3B2-Mediated RNA Splicing Drives Human Prostate Cancer Progression Norihiko Kawamura1,2, Keisuke Nimura1, Kotaro Saga1, Airi Ishibashi1, Koji Kitamura1,3, Hiromichi Nagano1, Yusuke Yoshikawa4, Kyoso Ishida1,5, Norio Nonomura2, Mitsuhiro Arisawa4, Jun Luo6, and Yasufumi Kaneda1 Abstract Androgen receptor splice variant-7 (AR-V7) is a General RNA splicing SF3B2 complex-mediated alternative RNA splicing constitutively active AR variant implicated in U2 castration-resistant prostate cancers. Here, we show U2 snRNA that the RNA splicing factor SF3B2, identified by 3’ 3’ in silico and CRISPR/Cas9 analyses, is a critical 5’ 3’ splice site 5’ SF3B7 AR-V7 5’ A U2AF2 AGA Exon ? determinant of expression and is correlated SF3B6(p14) SF3B4 SF3B1 SF3B4 SF3B1 with aggressive cancer phenotypes. Transcriptome SF3B5 SF3B2 SF3B3 SF3B2 SF3B3 and PAR-CLIP analyses revealed that SF3B2 con- SF3A3 SF3B2 complex SF3A3 SF3A1 SF3A1 SF3b complex trols the splicing of target genes, including AR, to AR pre-mRNA drive aggressive phenotypes. SF3B2-mediated CE3 aggressive phenotypes in vivo were reversed by AR-V7 mRNA AR mRNA AR-V7 knockout. Pladienolide B, an inhibitor of CE3 a splicing modulator of the SF3b complex, sup- Drive malignancy pressed the growth of tumors addicted to high While the SF3b complex is critical for general RNA splicing, SF3B2 promotes inclusion of the target exon through recognizing a specific RNA motif. SF3B2 expression. These findings support the idea © 2019 American Association for Cancer Research that alteration of the splicing pattern by high SF3B2 expression is one mechanism underlying prostate cancer progression and therapeutic resistance. -

Minor Intron Retention Drives Clonal Hematopoietic Disorders and Diverse Cancer Predisposition
ARTICLES https://doi.org/10.1038/s41588-021-00828-9 Minor intron retention drives clonal hematopoietic disorders and diverse cancer predisposition Daichi Inoue1,2,12, Jacob T. Polaski 3,12, Justin Taylor 4,12, Pau Castel 5, Sisi Chen2, Susumu Kobayashi1,6, Simon J. Hogg2, Yasutaka Hayashi1, Jose Mario Bello Pineda3,7,8, Ettaib El Marabti2, Caroline Erickson2, Katherine Knorr2, Miki Fukumoto1, Hiromi Yamazaki1, Atsushi Tanaka1,9, Chie Fukui1, Sydney X. Lu2, Benjamin H. Durham 2, Bo Liu2, Eric Wang2, Sanjoy Mehta10, Daniel Zakheim10, Ralph Garippa10, Alex Penson2, Guo-Liang Chew 11, Frank McCormick 5, Robert K. Bradley 3,7 ✉ and Omar Abdel-Wahab 2 ✉ Most eukaryotes harbor two distinct pre-mRNA splicing machineries: the major spliceosome, which removes >99% of introns, and the minor spliceosome, which removes rare, evolutionarily conserved introns. Although hypothesized to serve important regulatory functions, physiologic roles of the minor spliceosome are not well understood. For example, the minor spliceosome component ZRSR2 is subject to recurrent, leukemia-associated mutations, yet functional connections among minor introns, hematopoiesis and cancers are unclear. Here, we identify that impaired minor intron excision via ZRSR2 loss enhances hemato- poietic stem cell self-renewal. CRISPR screens mimicking nonsense-mediated decay of minor intron-containing mRNA species converged on LZTR1, a regulator of RAS-related GTPases. LZTR1 minor intron retention was also discovered in the RASopathy Noonan syndrome, due to intronic mutations disrupting splicing and diverse solid tumors. These data uncover minor intron rec- ognition as a regulator of hematopoiesis, noncoding mutations within minor introns as potential cancer drivers and links among ZRSR2 mutations, LZTR1 regulation and leukemias. -

Open Data for Differential Network Analysis in Glioma
International Journal of Molecular Sciences Article Open Data for Differential Network Analysis in Glioma , Claire Jean-Quartier * y , Fleur Jeanquartier y and Andreas Holzinger Holzinger Group HCI-KDD, Institute for Medical Informatics, Statistics and Documentation, Medical University Graz, Auenbruggerplatz 2/V, 8036 Graz, Austria; [email protected] (F.J.); [email protected] (A.H.) * Correspondence: [email protected] These authors contributed equally to this work. y Received: 27 October 2019; Accepted: 3 January 2020; Published: 15 January 2020 Abstract: The complexity of cancer diseases demands bioinformatic techniques and translational research based on big data and personalized medicine. Open data enables researchers to accelerate cancer studies, save resources and foster collaboration. Several tools and programming approaches are available for analyzing data, including annotation, clustering, comparison and extrapolation, merging, enrichment, functional association and statistics. We exploit openly available data via cancer gene expression analysis, we apply refinement as well as enrichment analysis via gene ontology and conclude with graph-based visualization of involved protein interaction networks as a basis for signaling. The different databases allowed for the construction of huge networks or specified ones consisting of high-confidence interactions only. Several genes associated to glioma were isolated via a network analysis from top hub nodes as well as from an outlier analysis. The latter approach highlights a mitogen-activated protein kinase next to a member of histondeacetylases and a protein phosphatase as genes uncommonly associated with glioma. Cluster analysis from top hub nodes lists several identified glioma-associated gene products to function within protein complexes, including epidermal growth factors as well as cell cycle proteins or RAS proto-oncogenes. -

Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features
cancers Review Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features Valeria Visconte 1, Megan O. Nakashima 2 and Heesun J. Rogers 2,* 1 Department of Translational Hematology and Oncology Research, Taussig Cancer Institute, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] 2 Department of Laboratory Medicine, Cleveland Clinic, Cleveland, OH 44195, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-216-445-2719 Received: 15 October 2019; Accepted: 19 November 2019; Published: 22 November 2019 Abstract: Components of the pre-messenger RNA splicing machinery are frequently mutated in myeloid malignancies. Mutations in LUC7L2, PRPF8, SF3B1, SRSF2, U2AF1, and ZRSR2 genes occur at various frequencies ranging between 40% and 85% in different subtypes of myelodysplastic syndrome (MDS) and 5% and 10% of acute myeloid leukemia (AML) and myeloproliferative neoplasms (MPNs). In some instances, splicing factor (SF) mutations have provided diagnostic utility and information on clinical outcomes as exemplified by SF3B1 mutations associated with increased ring sideroblasts (RS) in MDS-RS or MDS/MPN-RS with thrombocytosis. SF3B1 mutations are associated with better survival outcomes, while SRSF2 mutations are associated with a shorter survival time and increased AML progression, and U2AF1 mutations with a lower remission rate and shorter survival time. Beside the presence of mutations, transcriptomics technologies have shown that one third of genes in AML patients are differentially expressed, leading to altered transcript stability, interruption of protein function, and improper translation compared to those of healthy individuals. The detection of SF mutations demonstrates the importance of splicing abnormalities in the hematopoiesis of MDS and AML patients given the fact that abnormal splicing regulates the function of several transcriptional factors (PU.1, RUNX1, etc.) crucial in hematopoietic function. -

Based on Network Pharmacology and RNA Sequencing Techniques to Explore the Molecular Mechanism of Huatan Jiangzhuo Decoction for Treating Hyperlipidemia
Hindawi Evidence-Based Complementary and Alternative Medicine Volume 2021, Article ID 9863714, 16 pages https://doi.org/10.1155/2021/9863714 Research Article Based on Network Pharmacology and RNA Sequencing Techniques to Explore the Molecular Mechanism of Huatan Jiangzhuo Decoction for Treating Hyperlipidemia XiaowenZhou ,1 ZhenqianYan ,1 YaxinWang ,1 QiRen ,1 XiaoqiLiu ,2 GeFang ,3 Bin Wang ,4 and Xiantao Li 1 1Laboratory of TCM Syndrome Essence and Objectification, School of Basic Medical Sciences, Guangzhou University of Chinese Medicine, Guangzhou 510006, China 2Guangzhou Sagene Tech Co., Ltd., Guangzhou 510006, China 3College of Traditional Chinese Medicine, Hunan University of Chinese Medicine, Changsha 410208, China 4Shenzhen Traditional Chinese Medicine Hospital, Shenzhen 518000, China Correspondence should be addressed to Xiantao Li; [email protected] Received 22 May 2020; Revised 12 March 2021; Accepted 18 March 2021; Published 12 April 2021 Academic Editor: Jianbo Wan Copyright © 2021 Xiaowen Zhou et al. +is is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Background. Hyperlipidemia, due to the practice of unhealthy lifestyles of modern people, has been a disturbance to a large portion of population worldwide. Recently, several scholars have turned their attention to Chinese medicine (CM) to seek out a lipid-lowering approach with high efficiency and low toxicity. +is study aimed to explore the mechanism of Huatan Jiangzhuo decoction (HTJZD, a prescription of CM) in the treatment of hyperlipidemia and to determine the major regulation pathways and potential key targets involved in the treatment process. -
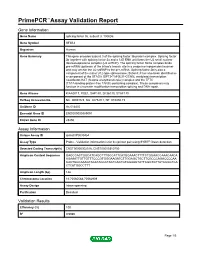
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name splicing factor 3b, subunit 3, 130kDa Gene Symbol SF3B3 Organism Human Gene Summary This gene encodes subunit 3 of the splicing factor 3b protein complex. Splicing factor 3b together with splicing factor 3a and a 12S RNA unit forms the U2 small nuclear ribonucleoproteins complex (U2 snRNP). The splicing factor 3b/3a complex binds pre-mRNA upstream of the intron's branch site in a sequence independent manner and may anchor the U2 snRNP to the pre-mRNA. Splicing factor 3b is also a component of the minor U12-type spliceosome. Subunit 3 has also been identified as a component of the STAGA (SPT3-TAF(II)31-GCN5L acetylase) transcription coactivator-HAT (histone acetyltransferase) complex and the TFTC (TATA-binding-protein-free TAF(II)-containing complex). These complexes may function in chromatin modification transcription splicing and DNA repair. Gene Aliases KIAA0017, RSE1, SAP130, SF3b130, STAF130 RefSeq Accession No. NC_000016.9, NG_027529.1, NT_010498.15 UniGene ID Hs.514435 Ensembl Gene ID ENSG00000189091 Entrez Gene ID 23450 Assay Information Unique Assay ID qHsaCIP0030454 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000302516, ENST00000310750 Amplicon Context Sequence GAGCCACTGGCATCAGCTTTGCCATTCATGGAAACTTTTCTGGAACCAAACAACA AGAAATTGTTGTTTCCCGTGGGAAGATCTTGGAGCTGCTTCGCCCAGACCCCAA CACTGGCAAAGTACATACCCTACTCACTGTGGAAGTATTCGGTGTTATCCGGTCA CTCATGGCCTTT Amplicon Length (bp) 146 Chromosome Location 16:70560588-70562909 Assay Design Intron-spanning Purification Desalted Validation Results Efficiency (%) 100 R2 0.9986 Page 1/5 PrimePCR™Assay Validation Report cDNA Cq 18.72 cDNA Tm (Celsius) 83 gDNA Cq Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

The Biological Function and Clinical Significance of SF3B1 Mutations In
Zhou et al. Biomarker Research (2020) 8:38 https://doi.org/10.1186/s40364-020-00220-5 REVIEW Open Access The biological function and clinical significance of SF3B1 mutations in cancer Zhixia Zhou1* , Qi Gong2, Yin Wang1, Mengkun Li1, Lu Wang1, Hongfei Ding1 and Peifeng Li1* Abstract Spliceosome mutations have become the most interesting mutations detected in human cancer in recent years. The spliceosome, a large, dynamic multimegadalton small nuclear ribonucleoprotein composed of small nuclear RNAs associated with proteins, is responsible for removing introns from precursor mRNA (premRNA) and generating mature, spliced mRNAs. SF3B1 is the largest subunit of the spliceosome factor 3b (SF3B) complex, which is a core component of spliceosomes. Recurrent somatic mutations in SF3B1 have been detected in human cancers, including hematological malignancies and solid tumors, and indicated to be related to patient prognosis. This review summarizes the research progress of SF3B1 mutations in cancer, including SF3B1 mutations in the HEAT domain, the multiple roles and aberrant splicing events of SF3B1 mutations in the pathogenesis of tumors, and changes in mutated cancer cells regarding sensitivity to SF3B small-molecule inhibitors. In addition, the potential of SF3B1 or its mutations to serve as biomarkers or therapeutic targets in cancer is discussed. The accumulated knowledge about SF3B1 mutations in cancer provides critical insight into the integral role the SF3B1 protein plays in mRNA splicing and suggests new targets for anticancer therapy. Keywords: SF3B1, Mutation, Cancer, RNA splicing Background human diseases, including cancer [5, 6]. Indeed, genome- Of the 3.3 billion base pairs of haploid DNA in the human wide studies have revealed more than 15,000 tumor- genome, approximately 20,000 protein-coding genes have associated splice variants in a wide variety of cancers [7, 8]. -

Dramatically Reduced Spliceosome in Cyanidioschyzon Merolae
Dramatically reduced spliceosome in PNAS PLUS Cyanidioschyzon merolae Martha R. Starka, Elizabeth A. Dunnb, William S. C. Dunna, Cameron J. Grisdalec, Anthony R. Danielea, Matthew R. G. Halsteada, Naomi M. Fastc, and Stephen D. Radera,b,1 aDepartment of Chemistry, University of Northern British Columbia, Prince George, BC, V2N 4Z9 Canada; and Departments of bBiochemistry and Molecular Biology, and cBotany, University of British Columbia, Vancouver, BC, V6T 1Z4 Canada Edited by Joan A. Steitz, Howard Hughes Medical Institute, Yale University, New Haven, CT, and approved February 9, 2015 (received for review September 1, 2014) The human spliceosome is a large ribonucleoprotein complex that C. merolae is an acidophilic, unicellular red alga that grows at catalyzes pre-mRNA splicing. It consists of five snRNAs and more temperatures of up to 56 °C (6). At 16.5 million base pairs, its than 200 proteins. Because of this complexity, much work has genome is similar in size to that of S. cerevisiae and contains focusedontheSaccharomyces cerevisiae spliceosome, viewed as a comparable number of genes; however only one tenth as many a highly simplified system with fewer than half as many splicing introns were annotated in C. merolae: 26 intron-containing factors as humans. Nevertheless, it has been difficult to ascribe genes, 0.5% of the genome (6). The small number of introns in a mechanistic function to individual splicing factors or even to dis- C. merolae raises the questions of whether the full complexity cern which are critical for catalyzing the splicing reaction. We have of the canonical splicing machinery has been maintained or C merolae identified and characterized the splicing machinery from the red alga whether .