The Carbonatation of Gypsum: Pathways and Pseudomorph Formation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Municipal Solid Waste Incineration (MSWI) Ashes for Greener Concrete Master’S Project for the Master Program of Structural
Municipal solid waste incineration (MSWI) ashes for greener concrete Master’s project for the Master Program of Structural Engineering and Building Technology ARNAUD GLIKSON SARA LÓPEZ MENÉNDEZ Department of Architecture and Civil Engineering Division of Building Technology Building Materials CHALMERS UNIVERSITY OF TECHNOLOGY Master’s Thesis ACEX30-19-97 Gothenburg, Sweden 2019 MASTER’S THESIS ACEX30-19-97 Municipal solid waste incineration (MSWI) ashes for greener concrete Master’s project for the Master Program of Structural Engineering and Building Technology ARNAUD GLIKSON SARA LÓPEZ MENÉNDEZ Department of Architecture and Civil Engineering Division of Building Technology Building Materials CHALMERS UNIVERSITY OF TECHNOLOGY Göteborg, Sweden 2019 Municipal solid waste incineration (MSWI) ashes for greener concrete Master’s project for the Master Program of Structural Engineering and Building Technology ARNAUD GLIKSON SARA LÓPEZ MENÉNDEZ © ARNAUD GLIKSON, SARA LÓPEZ MENÉNDEZ, 2019 Examensarbete ACEX30-19-97 Institutionen för arkitektur och samhällsbyggnadsteknik Chalmers tekniska högskola, 2019 Department of Architecture and Civil Engineering Division of Building Technology Building Materials Chalmers University of Technology SE-412 96 Göteborg Sweden Telephone: + 46 (0)31-772 1000 Department of Architecture and Civil Engineering Göteborg, Sweden, 2019 Municipal solid waste incineration (MSWI) ashes for greener concrete Master’s project for the Master Program of Structural Engineering and Building Technology ARNAUD GLIKSON SARA LÓPEZ MENÉNDEZ Department of Architecture and Civil Engineering Division of Building Technology Building Materials Chalmers University of Technology ABSTRACT Owing to the varying characteristics of municipal solid waste incineration (MSWI) ashes and the lack of harmonized standards and regulations, a substantial portion of MSWI ashes are simply used as landfilling cover at the present, which can be better utilized in the viewpoint of sustainability. -

Opportunities for Geologic Carbon Sequestration in Washington State
Opportunities for Geologic Carbon Sequestration in Washington State Contributors: Jacob R. Childers, Ryan W. Daniels, Leo F. MacLeod, Jonathan D. Rowe, and Chrisopher R. Walker Faculty Advisor: Juliet G. Crider Department of Earth and Space Sciences University of Washington, Seattle May 2020 ESS Special Topics Task Force Report 001 Preface and Acknowledgment This report is the product of reading, conversation, writing and revision by a group of undergraduate student authors during 10 weeks of March, April and May 2020. Our intent is to review the basic processes and state of current scientific understanding of geologic carbon sequestration relevant to Washington State. We would like to thank Dr. Thomas L. Doe (Golder Associates) for reading the final report and asking us challenging questions. i I. Introduction Anthropogenic emissions of greenhouse gasses like CO2 are raising global temperatures at an unprecedented rate. According to the most recent United Nations Intergovernmental Panel on Climate Change report, global emissions have caused ~1°C global warming above pre-industrial levels, with impacts such as rising sea level and changing weather patterns (IPCC 2018). The IPCC states that the current rate of emissions will lead to global warming of 1.5°C sometime between 2030 to 2052, and that temperature will continue to rise above that if emissions are not halted. They state that impacts of global warming, such as drought or extreme precipitation, will increase with a 1.5°C temperature increase. However, these effects will be less than the impacts of global temperature increase of beyond 2°C. The IPCC also projects that global sea level rise will be 0.1 m lower at 1.5°C warming than at 2°C warming, exposing 10 million fewer people to risks related to sea level rise. -

The New IMA List of Gem Materials – a Work in Progress – Updated: July 2018
The New IMA List of Gem Materials – A Work in Progress – Updated: July 2018 In the following pages of this document a comprehensive list of gem materials is presented. The list is distributed (for terms and conditions see below) via the web site of the Commission on Gem Materials of the International Mineralogical Association. The list will be updated on a regular basis. Mineral names and formulae are from the IMA List of Minerals: http://nrmima.nrm.se//IMA_Master_List_%282016-07%29.pdf. Where there is a discrepancy the IMA List of Minerals will take precedence. Explanation of column headings: IMA status: A = approved (it applies to minerals approved after the establishment of the IMA in 1958); G = grandfathered (it applies to minerals discovered before the birth of IMA, and generally considered as valid species); Rd = redefined (it applies to existing minerals which were redefined during the IMA era); Rn = renamed (it applies to existing minerals which were renamed during the IMA era); Q = questionable (it applies to poorly characterized minerals, whose validity could be doubtful). Gem material name: minerals are normal text; non-minerals are bold; rocks are all caps; organics and glasses are italicized. Caveat (IMPORTANT): inevitably there will be mistakes in a list of this type. We will be grateful to all those who will point out errors of any kind, including typos. Please email your corrections to [email protected]. Acknowledgments: The following persons, listed in alphabetic order, gave their contribution to the building and the update of the IMA List of Minerals: Vladimir Bermanec, Emmanuel Fritsch, Lee A. -

The Telephone City Crystal Brantford Lapidary & Mineral Society
THE TELEPHONE CITY CRYSTAL BRANTFORD LAPIDARY & MINERAL SOCIETY JANUARY 2017 Volume 71 Issue 1 INSIDE THIS ISSUE: JANUARY MEETING— 2 EARTH ON THE MOVE& ARAGONITE UPCOMING EVENTS & 3 CLUB INFO PIECE OF DINOSAUR 4 TAIL IN AMBER RARE GEM MINE 5 & TUMBLER GRIT ROCK TUMBLING 6 MONTHLY MINERAL - 7 APATITE 2016 EXECUTIVE & 8 MISC. December Meeting Highlight Photo- listing on page 2 1 THE TELEPHONE CITY CRYSTAL DATE: FRIDAY JANUARY 20, 2017 TIME: 7:30 PM WHERE: TB COSTAIN/S.C. JOHNSON COMMUNITY CENTER, 16 MORRELL ST. BRANTFORD, ONT. PROGRAM: RENE PERRIN: “EARTH ON THE MOVE” “The title is "Earth on the Move" featuring how the earth has moved and is mov- ing now at both the large scale as in India moving North with various other big and much smaller ex- amples with a beautiful graphic showing all of the global movements as tracked by stations all over the world. This is a great image showing the movement directions with arrows.” THE MINERAL ARAGONITE Calcium carbonate forms as both Aragonite and Calcite, and these two minerals only differ in theircrystallization. Cal- cite, the more common mineral, forms in trigonal crystals, whereas Aragonite forms orthorhombic crystals. On occasion, crystals of Aragonite and Calcite are too small to be individually determined, and it is only possible to distinguish these two minerals with optical or x-ray testing. The true identity of microcrystalline forms of Aragonite or Calcite may also not be known without complex testing, and this can also cause a confusion between these species. Most large Aragonite crystals are twinned growths of three individual crystals that form pseudohexagonal trillings. -

BOOK of ABSTRACTS SCK•CEN-BA-53 13/Dja/P-23
BOOK OF ABSTRACTS SCK•CEN-BA-53 13/Dja/P-23 3rd International Workshop Mechanisms and modelling of waste/cement interactions Ghent, 06-08 May 2013 SCK•CEN Boeretang 200 BE-2400 MOL Belgium http://www.sckcen.be BOOK OF ABSTRACTS SCK•CEN-BA-53 3rd International Workshop Mechanisms and modelling of waste/cement interactions Ghent, 06-08 May 2013 SCK•CEN, Boeretang 200, BE-2400 MOL, Belgium Contact: Diederik Jacques Tel: +32 14 33 32 09 E-mail: [email protected] Organising committee Workshop convenors Geert De Schutter, UGent, B Diederik Jacques, SCK•CEN, B Hans Meeussen, NRG, NL Hans van der Sloot, ECN, NL Tom van Gerven, KU Leuven, B Lian Wang, SCK•CEN, B International Steering Committee Urs Berner, PSI, CH Céline Cau Dit Coumes, CEA, F Frederic Glasser, Aberdeen University, UK Bernd Grambow, SUBATECH, F James Kirkpatrick, Michigan State University, USA Barbara Lothenbach, Empa, CH André Nonat, Univ. Bourgogne, F © SCK•CEN Studiecentrum voor Kernenergie Centre d’Etude de l’Energie Nucléaire Boeretang 200 BE-2400 MOL Belgium http://www.sckcen.be Status: Unclassified RESTRICTED All property rights and copyright are reserved. Any communication or reproduction of this document, and any communication or use of its content without explicit authorization is prohibited. Any infringement to this rule is illegal and entitles to claim damages from the infringer, without prejudice to any other right in case of granting a patent or registration in the field of intellectual property. Copyright of the individual abstracts and papers rests with the authors and the SCK•CEN takes no responsability on behalf of their content and their further use. -
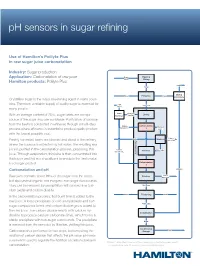
Ph Sensors in Sugar Refining
pH sensors in sugar refining Use of Hamilton’s Polilyte Plus in raw sugar juice carbonatation Industry: Sugar production Application: Carbonatation of raw juice Sugar Washing Beets Slicing Hamilton products: Polilyte Plus Pellets Slices Drying Water Extraction Scraps Pelletation Crystalline sugar is the major sweetening agent in many coun- Lime tries. Therefore, a reliable supply of quality sugar is essential for Raw Juice CaCO3 many people. Lime Lime milk Ca(OH) Liming With an average content of 20%, sugar beets are a major burning 2 source of the sugar sucrose worldwide. Purification of sucrose from the beets is conducted in refineries through a multi-step Carbon Carbonatation Washing dioxide CO process where efficiency is essential to produce quality product 2 1 water with the lowest possible cost. Lime + Water Washing Freshly harvested beets are cleaned and sliced at the refinery, impurities g where the sucrose is extracted by hot water. The resulting raw Cleanin juice is purified in the carbonatation process, producing thin Carbon Filtration Filter cake dioxide CO2 juice. Through evaporation, thin juice is then concentrated into Juice thick juice and fed into crystallizers to produce the final crystal- Carbonatation lized sugar product. 2 Carbonatation and pH Melasse Lime + Raw juice contains about 99% of the sugar from the beets, Filtration impurities but also several organic and inorganic non-sugar compounds. They can be removed by precipitation with burned lime (cal- Thin Juice cium oxide) and carbon dioxide. Steam Thickening Steam In the carbonatation process, first burnt lime is added to the raw juice. A loose precipitate of calcium hydroxide and non- Thick Juice sugar compounds forms and carbon dioxide gas is added to the raw juice. -

Carbonation of Alkaline Paper Mill Waste to Reduce CO2 Greenhouse Gas Emissions Into the Atmosphere R
Carbonation of alkaline paper mill waste to reduce CO2 greenhouse gas emissions into the atmosphere R. Perez-Lopez, G. Montes-Hernandez, J.-M. Nieto, F. Renard, L. Charlet To cite this version: R. Perez-Lopez, G. Montes-Hernandez, J.-M. Nieto, F. Renard, L. Charlet. Carbonation of alkaline paper mill waste to reduce CO2 greenhouse gas emissions into the atmosphere. Applied Geochemistry, Elsevier, 2008, 23 (8), pp.2292 à 2300. 10.1016/j.apgeochem.2008.04.016. insu-00351929 HAL Id: insu-00351929 https://hal-insu.archives-ouvertes.fr/insu-00351929 Submitted on 12 Jan 2009 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. 1 Carbonation of alkaline paper mill waste to reduce CO2 greenhouse gas 2 emissions into the atmosphere 3 4 R. Pérez-López∗, a,b, G. Montes-Hernandez a, J.M. Nieto b, F. Renard c,d, L. Charlet a 5 6 a LGIT, CNRS-OSUG-UJF, Université Joseph Fourier, Grenoble I, Maison des Géosciences, BP 53, 7 38041 Grenoble Cedex, France 8 b Department of Geology, University of Huelva, Campus ‘El Carmen’, 21071, Huelva, Spain 9 c LGCA, CNRS-OSUG-UJF, Université Joseph Fourier, Grenoble I, Maison des Géosciences, BP 53, 10 38041 Grenoble Cedex, France 11 d Physics of Geological Processes, University of Oslo, Norway 12 13 14 To be submitted to Applied Geochemistry on December 18th, 2007 15 ∗ Corresponding author. -

May 16, 1967 W. F. WALDECK 3,320,0 6 METHOD of PREPARING CALCITE and the PRODUCT THEREOF Filed Feb
May 16, 1967 w. F. WALDECK 3,320,0 6 METHOD OF PREPARING CALCITE AND THE PRODUCT THEREOF Filed Feb. 20, 1964 5 Sheets-Sheet 1 PRIOR ART (.7, I____: ONE MICRON May 15, 1967 w. F. wALDEcK “ 3,320,026 METHOD OF PREPARING CALCITE vAND THE PRODUCT THEREOF Filed Feb. 20, 1964 5 Sheets-Sheet 2 5%, w M? l__:__.| ONE MICRON May 16, 1967 w. F. WALDECK 3,320,026 METHOD OF PREPARING CALCITE AND THE PRODUCT THEREOF Filed Feb. 20, 1964 5 Sheets-Sheet 5 O.l MICRON Fig.6 3,320,026 United States Patent nice Patented May 16, 1967 1 2 complete reaction to product substantially free of other 3,320,026 crystalline forms. METHDD 0F PREPARING CALCITE AND THE PRODUCT THEREOF Carbonatation may be conducted in standard gas-liquid William F. Waldeclr, Milford, N.J., assignor to Chas. contacting equipment. For example, an agitated tank P?zer & Co., Inc., New York, N.Y., a corporation of may be employed, carbon dioxide-containing gas being Delaware suitably injected beneath the agitator for thorough dis Filed Feb. 20, 1964, Ser. No. 346,349 persion throughout the reaction mixture. A typical plant 16 Claims. (Cl. 23-66) scale precipitation may require one or two hours, under which circumstances it will usually be appropriate to This invention is concerned with calcium carbonate, 10 maintain the temperature below 20° C. for about 10 or and more particularly with a calcite of novel crystal 15 minutes, by recirculating some of the slurry through habit, and processes for its preparation and use. -

Turquoise in the Burro Mountains, New Mexico
TURQUOISE IN THE BURRO MOUNTAINS, NEW MEXICO. EDWARD R. ZALINSKI. [The following notes are from data collected during the year •9o5 while the writer was superintendent of the Azure Mining Company's property near Silver City. It was his intention to make a detailed study of the sub- ject, but this professional duties have hitherto prevented and in the following pages are presented the tentative conclusionsof an unfinished investigation.] HISTORY AND LOCALITIES. Turquoiseoriginally came fr4m' Persia by wayof Turkeyand was importedby the Venetians,who called it Turchesa,of which its presentname is the French variation. In America it has long been known to the southwesternIndians and was used by them for ornaments and mosaic work. Professor W. P. Blake, in •858 and '59, called attention to its occurrence at Cerrillos, New Mexico, where it had been mined by the aboriginesand early Spaniards. This locality has producedsome valuable gems, but the mines are not at present worked. Turquoise has been found also at Turquoise Moun- tain, CochiseCounty, and Mineral Park, Mohave County, Ari- zona; near Columbus and near Crescent, in southern Nevada; in Fresno County, California; and in Colorado. In New Mexico, besides Cerrillos mentioned above, it is known at Hachiti; in the Burro Mountains, Grant County; and in the Jarrilla Moun- tains, Otero County. The modern discovery of turquoise in the Burro Mountains dates from •875, but these depositswere known to the Indians and were worked by them. Remains of ancient operationsare still to be seen, while stone hammers, implements and fragments of pottery have been found near the old excavations. -

CO2 Sequestration in the Production of Portland Cement Mortars with Calcium Carbonate Additions
nanomaterials Article CO2 Sequestration in the Production of Portland Cement Mortars with Calcium Carbonate Additions Marius-George Parvan, Georgeta Voicu *, Alina-Ioana Badanoiu, Adrian-Ionut Nicoara and Eugeniu Vasile Department of Science and Engineering of Oxide Materials and Nanomaterials, Faculty of Applied Chemistry and Material Science, University POLITEHNICA of Bucharest, 1-7 Gh. Polizu Street, District 1, RO-011061 Bucharest, Romania; [email protected] (M.-G.P.); [email protected] (A.-I.B.); [email protected] (A.-I.N.); [email protected] (E.V.) * Correspondence: [email protected] Abstract: The paper presents the obtention and characterization of Portland cement mortars with limestone filler and nano-calcite additions. The nano-calcite was obtained by the injection of CO2 in a nano-Ca(OH)2 suspension. The resulted nano-CaCO3 presents different morphologies, i.e., polyhedral and needle like crystals, depending on the initial Ca(OH)2 concentration of the sus- pension. The formation of calcium carbonate in suspensions was confirmed by X-ray diffraction (XRD), complex thermal analysis (DTA-TG), scanning electron microscopy (SEM) and transmis- sion electron microscopy (TEM and HRTEM). This demonstrates the viability of this method to successfully sequestrate CO2 in cement-based materials. The use of this type of nano-CaCO3 in mortar formulations based on PC does not adversely modify the initial and final setting time of cements; for all studied pastes, the setting time decreases with increase of calcium carbonate content (irrespective of the particle size). Specific hydrated phases formed by Portland cement hydration Citation: Parvan, M.-G.; Voicu, G.; Badanoiu, A.-I.; Nicoara, A.-I.; Vasile, were observed in all mortars, with limestone filler additions or nano-CaCO3, irrespective of curing E. -

WITWATERSRAND GEM and MINERAL CLUB
WITWATERSRAND GEM and MINERAL CLUB Newsletter for SEPTEMBER 2014 P O Box 3708 Cresta 2118 South Africa nd Room 215 G, 2 floor Department of Geology University of Johannesburg Auckland Park JUST A WORD FROM THE EDITOR Whilst we are surviving hopefully the last of the winter, spring is upon us. And so out with the old and in with the new - the new amended constitution was accepted on the 13th August at the Special General Meeting. Thank you for all who attended the meeting, voted and enjoyed the social evening afterwards. Please forward any gem or mineral news, adverts or articles which you feel should be within our next newsletter, please forward to [email protected] or call me on 072 591 6202. Rock on!!! EVENT CALENDAR FOR 2014 – Please diarize the following dates THEMED EVENING on 10th September at UJ – Bring specimens relating to the Feldspar Group and Pseudomorphs SPEAKER EVENING on 24th September at UJ – no speaker due to the public holiday, however we will view a presentation from the Dallas Mineral Collection Symposium, 2013 The 21st General Meeting of the International Mineralogical Association will be held in South Africa at the Sandton Convention Centre, Gauteng, from 1–5 September 2014. For more information go to www.ima2014.co.za Kimberley Diamond Symposium & Trade Show: 11 - 13 September 2014 African Exploration Showcase : 7 November 2014 WGMC Year-end evening: 26 November 2014 (members only) CLUB MEETINGS The club meets every second and last Wednesday of each month at 19h00 for 19h30, excluding December, at the University of Johannesburg, Auckland Park, Department of Geology, Second Floor. -

Investigation Into the Filterability of Raw Sugars from Different Geographical Regions of the World
International Journal of Computational and Theoretical Chemistry 2017; 5(6): 53-58 http://www.sciencepublishinggroup.com/j/ijctc doi: 10.11648/j.ijctc.20170506.11 ISSN: 2376-7286 (Print); ISSN: 2376-7308 (Online) Investigation into the Filterability of Raw Sugars from Different Geographical Regions of the World Kamal Suleiman Hassan1, Kanada Toto Korea2, Abubakr Musa1, 3, * 1Sugar Institute, University of Gezira, Wad Madani, Algezira, Sudan 2Alkhaleej Sugar Co., Dubai, UAE 3School of Food and Biological Engineering, Jiangsu University, Zhenjiang, China Email address: [email protected] (A. Musa) *Corresponding author To cite this article: Kamal Suleiman Hassan, Kanada Toto Korea, Abubakr Musa. Investigation into the Filterability of Raw Sugars from Different Geographical Regions of the World. International Journal of Computational and Theoretical Chemistry. Vol. 5, No. 6, 2017, pp. 53-58. doi: 10.11648/j.ijctc.20170506.11 Received: October 19, 2017; Accepted: November 8, 2017; Published: January 2, 2018 Abstract: This study was carried out at Alkhaleej Sugar Refinery in Dubai, United Arab Emirates. The study aimed to find out the correlation between the filterability and slurry resistance value so fraw meltliquor in the carbonation sugar refinery and the raw sugar quality. The investigation utilized 5 raw sugar samples originated from various regions of the world comprising three quality groups namely; low pol sugar (LP) originated from Thailand, very high pol sugar (VHP) originated from Brazil, South-Africa, Sudan, India, and very high pol sugar (VVHP) originated from Brazil. The filterability and the slurry resistance tests were used as the main determining factors for the evaluation. The filterability of refined sugar (consideredas100%) was used as a reference value for comparison.