1960 May, 1964
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Cy Martin Collection
University of Oklahoma Libraries Western History Collections Cy Martin Collection Martin, Cy (1919–1980). Papers, 1966–1975. 2.33 feet. Author. Manuscripts (1968) of “Your Horoscope,” children’s stories, and books (1973–1975), all written by Martin; magazines (1966–1975), some containing stories by Martin; and biographical information on Cy Martin, who wrote under the pen name of William Stillman Keezer. _________________ Box 1 Real West: May 1966, January 1967, January 1968, April 1968, May 1968, June 1968, May 1969, June 1969, November 1969, May 1972, September 1972, December 1972, February 1973, March 1973, April 1973, June 1973. Real West (annual): 1970, 1972. Frontier West: February 1970, April 1970, June1970. True Frontier: December 1971. Outlaws of the Old West: October 1972. Mental Health and Human Behavior (3rd ed.) by William S. Keezer. The History of Astrology by Zolar. Box 2 Folder: 1. Workbook and experiments in physiological psychology. 2. Workbook for physiological psychology. 3. Cagliostro history. 4. Biographical notes on W.S. Keezer (pen name Cy Martin). 5. Miscellaneous stories (one by Venerable Ancestor Zerkee, others by Grandpa Doc). Real West: December 1969, February 1970, March 1970, May 1970, September 1970, October 1970, November 1970, December 1970, January 1971, May 1971, August 1971, December 1971, January 1972, February 1972. True Frontier: May 1969, September 1970, July 1971. Frontier Times: January 1969. Great West: December 1972. Real Frontier: April 1971. Box 3 Ford Times: February 1968. Popular Medicine: February 1968, December 1968, January 1971. Western Digest: November 1969 (2 copies). Golden West: March 1965, January 1965, May 1965 July 1965, September 1965, January 1966, March 1966, May 1966, September 1970, September 1970 (partial), July 1972, August 1972, November 1972, December 1972, December 1973. -

Download DECEMBER 1964.Pdf
Vol. 33, No. 12 December 1964 Federal Bureau of Investigation United States Department of Justice J. Edgar Hoover, Director Index to l'olume 33, 1964 (p. 27) Contents 1 Message from Director J. Edgar Hoover Feature Article: 3 Recruiting and Training of Police Personnel, by Joseph T. Carroll, Chief of Police, Lincoln, Nebr. FBI National Academy: 9 Marine Commandant, Noted Editor Address Graduates Scientific Aids:· 13 BuildingMaterial Evidence in Burglary Cases Nationwide Crimescope: 17 A 2 Gauge Cane 17 From "Pen" to "Sword" Vol. 33, No. 12 Crime Prevention: 18 A Mess<'toe for Young People, by Edward K. Dabrowski, Sheriff of Bri tol County, New Bedford, Mass. Other Topics: 26 Wanted by the FBI 27 Index to Articles Published During 1964 Publi.hed by the FEDERAL BUREAU Identification: OF INVESTIGATION, Questionable Pattern (back cover) UNITED STATES DEPARTMENT OF JUSTICE Wa.hlngton, D.C. 20535 MESSAGE FROM THE DIRECTOR TO ALL LAW E FORCEMENT OFFICIALS ATHEISTIC COMMUNISM and the lawless underworld are not the only threats to the safety and welfare of our great Nation. Enemies of freedom come under many guises. Our society today is in a great state of unrest. Many citizens are confused and troubled. For the first time, some are confronted with issues and decisions relating to the rights and dignity of their fellow countrymen, problems which heretofore they had skirted or ignored. We have in our midst hatemongers, bigots, and riotous agitators, many of whom are at opposite poles philosophically but who spew similar doctrines of prejudice and intolerance. They exploit hate and fear for personal gain and selfaggrandizement. -

Median and Average Sales Prices of New Homes Sold in United States
Median and Average Sales Prices of New Homes Sold in United States Period Median Average Jan 1963 $17,200 (NA) Feb 1963 $17,700 (NA) Mar 1963 $18,200 (NA) Apr 1963 $18,200 (NA) May 1963 $17,500 (NA) Jun 1963 $18,000 (NA) Jul 1963 $18,400 (NA) Aug 1963 $17,800 (NA) Sep 1963 $17,900 (NA) Oct 1963 $17,600 (NA) Nov 1963 $18,400 (NA) Dec 1963 $18,700 (NA) Jan 1964 $17,800 (NA) Feb 1964 $18,000 (NA) Mar 1964 $19,000 (NA) Apr 1964 $18,800 (NA) May 1964 $19,300 (NA) Jun 1964 $18,800 (NA) Jul 1964 $19,100 (NA) Aug 1964 $18,900 (NA) Sep 1964 $18,900 (NA) Oct 1964 $18,900 (NA) Nov 1964 $19,300 (NA) Dec 1964 $21,000 (NA) Jan 1965 $20,700 (NA) Feb 1965 $20,400 (NA) Mar 1965 $19,800 (NA) Apr 1965 $19,900 (NA) May 1965 $19,600 (NA) Jun 1965 $19,800 (NA) Jul 1965 $21,000 (NA) Aug 1965 $20,200 (NA) Sep 1965 $19,600 (NA) Oct 1965 $19,900 (NA) Nov 1965 $20,600 (NA) Dec 1965 $20,300 (NA) Jan 1966 $21,200 (NA) Feb 1966 $20,900 (NA) Mar 1966 $20,800 (NA) Apr 1966 $23,000 (NA) May 1966 $22,300 (NA) Jun 1966 $21,200 (NA) Jul 1966 $21,800 (NA) Aug 1966 $20,700 (NA) Sep 1966 $22,200 (NA) Oct 1966 $20,800 (NA) Nov 1966 $21,700 (NA) Dec 1966 $21,700 (NA) Jan 1967 $22,200 (NA) Page 1 of 13 Median and Average Sales Prices of New Homes Sold in United States Period Median Average Feb 1967 $22,400 (NA) Mar 1967 $22,400 (NA) Apr 1967 $22,300 (NA) May 1967 $23,700 (NA) Jun 1967 $23,900 (NA) Jul 1967 $23,300 (NA) Aug 1967 $21,700 (NA) Sep 1967 $22,800 (NA) Oct 1967 $22,300 (NA) Nov 1967 $23,100 (NA) Dec 1967 $22,200 (NA) Jan 1968 $23,400 (NA) Feb 1968 $23,500 (NA) Mar 1968 -

May June 1964 #157
President Whitman Reelected ESTERN PACIFIC'S board of di proximated $6,160,000, and that $1,- ilepoSls W rectors, in a meeting following 839,000 was being spent to rebuild 200 the annual shareholders meeting held box cars. Volume XVI , No. 3 MAY - JUNE, 1964 *Milepost No. 157 in San Francisco on June 24, reelected W. C. Brunberg, vice president Frederic B. Whitman as president for marketing, said that although first the next year. Other officers reelected quarter revenues were disappointing, Public Relations Department by the board were M. M. Christy, ex recent traffic movements, future traf ecutive vice president and general WESTERN PACIFIC RAILROAD fic indicators, and the overall economy manager; W. C. Brunberg, vice presi SACRAMENTO NORTHERN RY. indicate a substantially improved rev TIDEWATER SOUTHERN RY. dent-marketing; E. L. Van Dellen, vice enue picture for the second half of 526 Mission Street president and genel'al counsel; and 1964. San Francisco, Calif. 94105 Logan Paine, secretary. It was pointed out by M. M. Christy, Lee " Flash" Sherwood, Editor The board also approved organiza executive vice president and general tion changes which are reported on manager, that the Interstate Com Page 4. merce Commission's 1963 report of op The directors also declared a regular erating statistics of large railroads, quarterly dividend of 451 per share, ranked Western Pacific first nation payable August 17, 1964 to sharehold ally in average speed of freight trains *Mile post No. 157: ers of record August 3. at 29.5 miles per hour, and that our Roadmaster R. J. At the annual shareholders' meeting railroad was first in the Central West Mounkes and Anisian. -

SEC News Digest, 06-24-1964
SECURITIES AND EXCHANGE COMMISSION ~!E~~ IDIl@!E~~ .~. A brief summary of financial proposals filed with and actions by the S.E.C. ft'Cb~ (In ord.rlng f,,11 t.xt of R.I.a ••• from Publication. Unit, cit. numb.r) Washington 25. D.C. (Issue No. 64-6-18) FOR RELEASE _J.;...u...;,;n..;;.,e_2;",.,4......,...;1;,;;.9.:;".64'-_ - HERCULES POWDER FILES EXCHANGE PLAN. Hercules Powder Company, 910 Market St., Wilmington, Del., filed a registration statement (File 2~22537) with the SEC on June 22 seeking registration of 430,971 shares of $1.65 cumulative convertible Class A stock, to be issued in connection with the company's acquisition of all assets and liabilities of Haveg Industries, Inc. The shares are to be offered to Haveg stockholders in ex- change for all of Haveg's outstanding stock, at the rate of two of such Class A shares for each five common shares of Haveg. The company is engaged in manufacturing and selling a diversified line of chemicals and allied products. Upon the acquisition of Haveg, it will add to its present operations the conversion of plastic materials into engineered and custom fabricated products and the processing of other basic chemicals for industrial and government use. In addition to preferred and convertible Class A stock, the company has outstanding 18,303,362 shares of common stock. Henry A. Thouron is president and chairman of the executive committee. WEYERHAEUSER FILES STOCK PLANS. Weyerhaeuser Company, Tacoma Bldg., Tacoma, Wash., filed a registration statement (File 2-22538) with the SEC on June 22 seeking registration of 682,292 shares of stock, to be offered under its Incentive Stock Option Plan and 1964 Incentive Stock Option Plan. -

November 3, 1964 Issue (Dig110364.Pdf)
SECURITIES AND EXCHANGE COMMISSION i~JlW~ IDU@JI~tr , A brief summary of fincmciol prQpOSCIls filed with and actions by the S.E.e. Washington 25, D.C. (In .,eI.r"" .... t... .f ••1..... fro. ,,,lIlIc.t'... Ulllt, cit. II".II.r) I (Issue Ro. 64-11-2) FOR RELEASE Rovember 3. 1964 FIRST AMIItICAR TITLE IRS. "LIS FOI OFFERIRG AlO) SECONDARY. First Aaerican Title lnauranc:e & fruit Company. 421 R. ~in St., Santa Ana. Calif., filed a regiatration state.-nt (File 2-22892) with the SEC on :November 2 seeking regiatration of 250,107 sharea of capital stock. Of these shares, 175,107 are to be offered for public sale by the preaent bolders thereof aDd 75,000 by the cOlllpany. The offering is to b... de through underwriters h.aded by Dean Witter & Co., 632-4 S. Sprinl St., Loa Anleles. The public offerinl price ($17 per ahare 8I8Xt..*) and uDclerwritinl terms are to be supplied by aaemt.ent. The ca.pany is enl&led principally in the title insurance business and related activities. Ret proceeds from ita aale of additional atock will be used to increase working capital and to pay portiona of outstanding notes (agarel&ting $666,723) issued in connection with acquiaition of interesta in 14 title insurance com- paniea since 1957. The company has outstandinl 768,516 common shares, as adjusted to reflect a 3-for-l aplit to be effected in Rove"r. Management officials as a Iroup own 331 of the outstaDding stock. The prospec- tus lists 14 selling stockholders, including Christiana Oil Corp. -

October 29, 1964 Issue (Dig102964.Pdf)
St\.-UKlllt:t ANut tJ{l.tiANut \,;,utMMI:i:sh.J'N i1mw~ IDU~~~~ ~brief summary of financial proposals filed with and actions by the S.E.C. Washington 25, D.C. (In .,tI.rln, .... t.xt .f R.I ••••• fro.. Pullllc.t' ••• Unit, cit•• ".It.r) (Issue No. 64-10-20) FOR RELEASE .--:0:,;:c:.,:to.;,:b:.,:e;o;.,r...;:2:.:;,9.....-=1~964-.:- _ MISSISSIPPI P6L SEIlS ORDER. Mi.si •• ippi Power & Lisbt eo.pany, a public-utility .ub.idiary of Middle South Utilities. Inc.. baa applied to the SEC for an order under the Holding Company Act with re.pect to a propo.ed transfer of a portion of it. earned .urp1us to it. capital .tock account; and the eo..1a.ion has i••ued an order (Release 35-15142) givins intere.ted per.oDS until November 23 to reque.t a bearing thereon. According to the application, Mi •• i•• ippi propo.e. to tran.fer $2,850,000 from it. earned .urplu. account, which &aOunted to $9,010,108 On August 31, 1964, to it. common capital .tock account, thereby increasing the latter to $45,600,000. 'lEL-A-SIGH FILlS FOR SECONDAllY. Te1-A-Sip. Inc •• 3401 W. 47th se ,; Chicago, filed a reiistration .tat... nt (Ftle 2-22880) with the SEC on October 28 .eekins regi.tration of 89,635 out.tanding .hares of CoaaDn .tock.' The .hares are to be offered for public sale by the pre.ent holder. thereof from time to time on the Aaerican Stock Exchanl8 or in the over-the-counter market, at price. -

Almanac, May 1964, Vol. 10, No. 9
Proposal for Two-Year Experiment in Honor System is Presented An experimental Honor System, to remain in effect for two years, will be instituted in the undergraduate schools of the University in September, 1964, if it wins the final ap- Administration Statement proval of the various school faculties this month. Texts of the proposal were circulated to members of the On University Professorships the Committee of Deans, of faculty by Undergraduate The statement on the and criteria for which Dean Willis Winn of the Wharton School is chair- following purposes Professorships has been adopted by the Univer- man, on April 21. The proposal itself was drawn up, after University sity administration, according to Provost David R. Goddard: extensive discussion, by the Undergraduate Affairs Com- mittee of the University Council. "A limited group of the distinguished members of the shall be the as "The reliable determination of is to faculty designated by Trustees University only feasibility try Professors. A Professor shall be an eminent an Honor System experimentally for a reasonable period University on a basis," the committee said in its scholar who has demonstrated a breadth of intellectual voluntary submitting vision which several of the arts, sci- proposal. "Our aim is to substitute experience for spec- encompasses aspects ences or professional fields. His published works and pro- ulation in determining the feasibility of establishing a fessional activities must have attained wide recognition permanent Honor System at Pennsylvania. The committee from well-known scholarly sources. suggests that the University, by taking an empirical ap- proach, will have ample opportunity for evaluation without "Candidates for a University Professorship may be nom- commitment beyond the experimental period. -
View This Page In
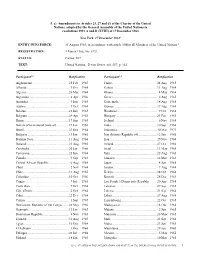
5. a) Amendments to Articles 23, 27 and 61 of the Charter of the United Nations, adopted by the General Assembly of the United Nations in resolutions 1991 A and B (XVIII) of 17 December 1963 New York, 17 December 19631 ENTRY. INTO FORCE: 31 August 1965, in accordance with article 108for all Members of the United Nations.2 REGISTRATION: 1 March 1966, No. 8132. STATUS: Parties: 107. TEXT: United Nations, Treaty Series, vol. 557, p. 143. Participant3,4 Ratification Participant3,4 Ratification Afghanistan..................................................25 Feb 1965 France ..........................................................24 Aug 1965 Albania......................................................... 7 Dec 1964 Gabon...........................................................11 Aug 1964 Algeria .........................................................26 Mar 1964 Ghana........................................................... 4 May 1964 Argentina ..................................................... 4 Apr 1966 Greece.......................................................... 2 Aug 1965 Australia....................................................... 9 Jun 1965 Guatemala....................................................18 Aug 1965 Austria ......................................................... 7 Oct 1964 Guinea..........................................................19 Aug 1964 Belarus.........................................................22 Jun 1965 Honduras...................................................... 9 Oct 1968 Belgium -

Power Reactors of the World
POWER REACTORS OF THE WORLD A. POWER REACTORS IN OPERATION, 1 Septembe r 1965 Name Location Type Net Output Criticality (MWe) Belgium BR-3 Mol Press. H20, 10.5 Aug 1962 3.7 + 4.4% U Canada NPD Rolphton Press. D20, 20 Apr 1962 nat. U France G-1 Marcoule Nan U, 3 Jan 1956 graphite, air G-2 (G-3) Marcoule Nat. U, 2 X 40 Jul 1958/ graphite, C02 Jun 1959 EDF-1 Chinon Nat. U, 70 Sep 1962 graphite, C02 EDF-2 Chinon Nat. U, 200 Aug 1964 graphite, C02 Germany, Federa • Republic of KAHL Gross we lzheim/ Boiling HjO 15 Nov I960 Kahl (Main) 2.6% U Italy LATINA Latin a Nat. U, 200 Dec 1962 (SIMEA) (Foce Verde) graphite, C02 GARIGLIANO Garigliano Boiling HjO 150 Jun 1963 (SENN) (Sessa Aurunca) 2% U ENRICO FERMI Trino Vercellese Press. HjO 186* Jun 1964 2.6 % U Japan JPDR Tokai-Mura Boiling H20 11.7 Aug 1963 2.5 % U TOKAI-MURA Tokai-Mura Nat. U 158 May 1965 graphite, C02 * to be raised to 257 MWe by the end of 1965. 26 Name Location Type Net Output Criticality (MWe) Sweden H-3/ADAM Agesta Press. D20 9 Jul 1963 nat. U United Kingdom CALDER HALL Calder Hall Nat. U 4 X 45 May 1956/ graphite, C02 Dec 1958 CHAPELCROSS Chapelcross Nau U 4 X 45 Oct 1958/ graphite, COa Dec 1959 DFR Dounreay Fast breeder 15 Nov 1959 45.5% U, NaK BERKELEY Berkeley Nau U 2 X 138 Aug 1961/ graphite, C02 Mar 1962 BRADWELL Bradwell Nat. -

United Nations Juridical Yearbook, 1964
Extract from: UNITED NATIONS JURIDICAL YEARBOOK 1964 Part Four. Legal documents index and bibliography of the United Nations and related intergovernmental organizations Chapter IX. Legal documents index of the United Nations and related intergovernmental organizations Copyright (c) United Nations CONTENTS (continued) Paç« Part Three. Judicial decisions on questions relating to the United Nations and related inter-governmental organizations CHAPTER VII. DECISIONS OF INTERNATIONAL TRIBUNALS 273 CHAPTER VIII. DECISIONS OF NATIONAL TRIBUNALS 1. Austria Highest Court, Austria Evangelical Church (Augsburg and Helvetic Confessions) v. Official of the IAEA: Judgement of 27 February 1964 Church dues are not taxes, but obligations under civil law—Article XV, section 38, of the Agreement regarding the Headquarters of the IAEA therefore does not grant exemption from the payment of church dues . 274 2. United States of America Westchester County Court Matter of foreclosure of tax liens by City of New Rochelle v. Republics of Ghana, Indonesia and Liberia: Judgement of 16 December 1964 Jurisdiction over proceedings to foreclose tax liens on residences of foreign representatives to the United Nations—Court declined to exercice juris- diction 275 Part Four. Legal documents index and bibliography of the United Nations and related inter-governmental organizations CHAPTER IX. LEGAL DOCUMENTS INDEX OF THE UNITED NATIONS AND RELATED INTER- GOVERNMENTAL ORGANIZATIONS A. LEGAL DOCUMENTS INDEX OF THE UNITED NATIONS 1. General Assembly and Subsidiary Organs 1. Plenary General Assembly and Main Committees Documents of legal interest 280 2. United Nations Conciliation Commission for Palestine Document of legal interest 281 3. Executive Committee of the Programme of the United Nations High Com- missioner for Refugees Documents of legal interest 282 4. -
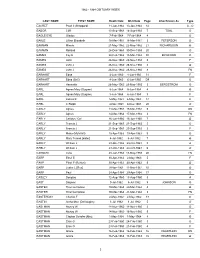
1960 to 1964 Volume 1: E-J Obituary Index
1960 - 1964 OBITUARY INDEX LAST NAME FIRST NAME Death Date Obit Date Page Also Known As Type EACRET Pearl I (Sheppard) 11-Jan-1964 13-Jan-1964 12 A - O EADOR Cliff 13-Sep-1963 14-Sep-1963 7 TOAL O EAGLE EYE Gladys 7-Feb-1964 7-Feb-1964 4 O EAKLE Lillian Elizabeth 18-Mar-1961 18-Mar-1961 3 PETERSON O EAKMAN Minnie 21-May-1962 22-May-1962 21 RICHARDSON O EAKMAN Rolland 26-Dec-1963 30-Dec-1963 20 O EAMES Fay E 28-Feb-1962 15-Mar-1962 30 BICKFORD O EAMES John 26-Nov-1960 29-Nov-1960 5 F EAMES John J 26-Nov-1960 26-Nov-1960 3 O EAMES John J 26-Nov-1960 28-Nov-1960 8 FN EARHART Edna 4-Jun-1960 8-Jun-1960 31 F EARHART Edna (Bell) 4-Jun-1960 6-Jun-1960 D9 O EARHART Henrietta 28-May-1963 29-May-1963 3 BERGSTROM O EARL Agnes Mary (Sugrne) 5-Jun-1964 5-Jun-1964 4 O EARL Agnes Mary (Sugrne) 5-Jun-1964 6-Jun-1964 5 F EARL James K 5-May-1964 6-May-1964 37 O EARL C Ralph 4-Nov-1963 4-Nov-1963 20 O EARLY Agnes 14-Mar-1964 16-Mar-1964 5 DN EARLY Agnes 14-Mar-1964 17-Mar-1964 3 FN EARLY Calistus "Cal' 16-Jun-1960 16-Jun-1960 7 O EARLY Francis J 21-Sep-1963 21-Sep-1963 5 O EARLY Francis J 21-Sep-1963 23-Sep-1963 3 F EARLY Marie (Mulvihill) 18-Apr-1963 19-Apr-1963 9 O EARLY Mary Teresa [sister] 4-Jul-1962 6-Jul-1962 7 O EARLY William J 23-Oct-1963 23-Oct-1963 5 A EARLY William J 23-Oct-1963 24-Oct-1963 6 O EARNEST Irene 28-Jan-1963 13-May-1963 20 FN EARP Ethel E 30-Apr-1963 2-May-1963 5 F EARP Ethel E (Burton) 30-Apr-1963 30-Apr-1963 5 O EARP Lester L [Rev] 9-Nov-1961 11-Nov-1961 10 O EARP Paul 28-Apr-1964 29-Apr-1964 37 O EASLEY Douglas 12-Aug-1960 13-Aug-1960