Case Study: Petroleum Origins of the Industry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Multicomponent Distillation
01/12/2009 Multicomponent distillation F. Grisafi 1 01/12/2009 Introduction The problem of determining the stage and reflux requitirements for multicomponen t distill a tions ismuch more complex than for binary mixtures. With a multicomponent mixture, fixing one component composition does not uniquely determine theother component compositions and the stage temperature. Also when the feed contains more than two components it is not possible to specify the complete composition of the top and bottom products independently. The separation between the top and bottom products is usually specified by setting limits on two "key components", between which it is desired to make the separation. 2 01/12/2009 Calculation procedure The normal procedure for a typical problem is to solve the MESH (Material balance, Equilibrium, Summation and Heat) balance equations stage-by-stage, from the top and bottom of the column toward the feed point. For such a calculation to be exact, the compositions obtained from both the bottom-up and top-down calculations must mesh at the feed point and mesh the feed composition. The calculated compositions will depend on the compositions assumed for the top and bottom products at the commencement of the calculations. Though it ispossibletomatch the key components, theother components will not match unless the designer was particularly fortunate in choosing the trial top and bottom compositions. 3 01/12/2009 Calculation procedure For a comppyletely rigorous solution the compositions must be adjusted and the calculations repeated until a satisfactory match at the feed point is obtained by iterative trial-and-error calculations. Clearly, the greater the number of components, the more difficult the problem. -

Exxonmobil Refinery Proposed Expansion: Operational Review and Neighborhood Demographic Profile
ExxonMobil Refinery Proposed Expansion: Operational Review and Neighborhood Demographic Profile Louisiana Democracy Project asked EBIC to examine the environmental track record of ExxonMobil’s Baton Rouge Refinery and also provide a demographic analysis of the neighborhoods within 2 miles of the Refinery. The ExxonMobil Refinery, previously knows as the Exxon Refinery, is located at 4045 Scenic Hwy. in Baton Rouge, LA. It borders the Mississippi River to the West and a densely populated neighborhood to the East with additional refineries extending to the north from its property boundary. The facility itself borders another Exxon Chemicals America plant (4999 Scenic Hwy.) which is also a significant source of toxic environmental releases, workplace hazards and regulatory problems. ExxonMobil is seeking a permit to expand its air pollution emissions at the Baton Rouge refinery. The company has claimed that even though the plant will produce more emissions, its overall operational effect on the environment would be make the air cleaner by producing a fuel that would lower automobile emissions. ExxonMobil is seeking permits to increase air pollution emissions at its Baton Rouge refinery. But, the company says the cleaner gasoline created by the project will mean fewer emissions from cars. The refinery wants to make changes so it can produce cleaner, low-sulfur gas as ordered by U.S. Environmental Protection Agency rules for 2004 model year cars and trucks. The new gasoline will mean that each year, vehicles in Baton Rouge's five-parish area will produce 2,238 fewer tons of nitrogen oxides and 842 fewer tons of volatile organic compounds. -

Clean Air Act (CAA) Watch List
Facilities on the Active May 2012 CAA Watch List The list of facilities below composes OECA's May 2012 Clean Air Act (CAA) Watch List. EPA maintains a "Watch List" to facilitate dialogue between EPA, state and local agencies on enforcement matters relating to facilities with alleged violations identified as either significant or high priority. Being on the Watch List may not mean that the facility has actually violated the law only that an evaluation or investigation by EPA or a state or local environmental agency has led those organizations to allege that an unproven violation has in fact occurred. The Watch List does not identify which alleged violations of environmental laws may pose the greatest risk to public health or the environment. It is an automated report based on data from the Air Facility System (AFS), which is used by federal, state and local agencies to track environmental enforcement and compliance information. Agencies input information into AFS, including information about violations that are identified as high priority violations (HPVs). Some facilities may appear on the Watch List due to data errors, which typically are identified and addressed during the EPA-state-local dialogue. EPA expects the government agency with jurisdiction over a facility with an HPV to initiate an appropriate enforcement response, in a timely manner, and input the action into AFS. Some facilities on the list are the subject of existing orders, are actively participating in negotiations with regulators, or are the subject of an investigation. While progress is being made toward resolving the violations, further activities may be required to achieve compliance. -

Safety Bulletin • Exxonmobil • Baton Rouge, Louisiana
Key Lessons from the ExxonMobil Baton Rouge Refinery Isobutane Release and Fire SUMMARY OF KEY LESSONS: Incident Date: November 22, 2016 Companies should: 4 Workers Injured • Evaluate human factors associated with No. 2016-02-I-LA equipment design and apply the hierarchy of controls to mitigate identified hazards. • Establish detailed and accurate written procedures and provide training to ensure workers can perform all anticipated job tasks safely. CSB • Safety Bulletin • ExxonMobil • Baton Rouge, Louisiana The U.S. Chemical Safety and Hazard Investigation Board (CSB) is an independent Federal agency whose mission is to drive chemical safety change through independent investigations to protect people and the environment. The CSB is a scientific investigative organization; it is not an enforcement or regulatory body. Established by the Clean Air Act Amendments of 1990, the CSB is responsible for determining accident causes, issuing safety recommendations, studying chemical safety issues, and evaluating the effectiveness of other government agencies involved in chemical safety. More information about the CSB is available at www.csb.gov. The CSB makes public its actions and decisions through investigative publications, all of which may include safety recommendations when appropriate. Examples of the types of publications include: CSB Investigation Reports: formal, detailed reports on significant chemical accidents and include key findings, root causes, and safety recommendations. CSB Investigation Digests: plain-language summaries of Investigation Reports. CSB Case Studies: examines fewer issues than a full investigative report, case studies present investigative information from specific accidents and include a discussion of relevant prevention practices. CSB Safety Bulletins: short, general-interest publications that provide new or timely information intended to facilitate the prevention of chemical accidents. -

Guide to the American Petroleum Institute Photograph and Film Collection, 1860S-1980S
Guide to the American Petroleum Institute Photograph and Film Collection, 1860s-1980s NMAH.AC.0711 Bob Ageton (volunteer) and Kelly Gaberlavage (intern), August 2004 and May 2006; supervised by Alison L. Oswald, archivist. August 2004 and May 2006 Archives Center, National Museum of American History P.O. Box 37012 Suite 1100, MRC 601 Washington, D.C. 20013-7012 [email protected] http://americanhistory.si.edu/archives Table of Contents Collection Overview ........................................................................................................ 1 Administrative Information .............................................................................................. 1 Arrangement..................................................................................................................... 3 Biographical / Historical.................................................................................................... 2 Scope and Contents........................................................................................................ 2 Names and Subjects ...................................................................................................... 4 Container Listing ............................................................................................................. 6 Series 1: Historical Photographs, 1850s-1950s....................................................... 6 Series 2: Modern Photographs, 1960s-1980s........................................................ 75 Series 3: Miscellaneous -

Example: Multicomponent Distillation with Shortcut Methods Consider A
2 Example: multicomponent distillation with shortcut methods Consider a single distillation column as shown in Fig. 6-1. Short-cut methods have been developed that allow you to see the splits that can be achieved at different pressures, with different reflux ratios, and with a different number of stages. It is often useful to do a short-cut distillation calculation first, before doing the more rigorous plate- to-plate calculations, because the preliminary calculations give you an idea of what is reasonable. The methods are based on the relative volatility of the chemicals, and require you to identify two components that will be essentially separated (to the extent you specify). If you line up all the components in the order of their boiling points, and draw a line between two of them, the more volatile component is called the light key and the less volatile component is called the heavy key. B1 Figure 6-1. Distillation Column For this example, you choose the first column in the process shown in Fig. 5-4. In Aspen Plus you use the module DSTWU for the shortcut method; you also use RK-Soave as the physical property method, because it is a good one for hydrocarbons. The feed is 100 lb. moles/hr of propane, 300 lb. moles/hr of i-butane, and the other chemicals as listed in Table 5-1 or Table 6-6, at 138 psia and 75 ºF. The column operates at 138 psia with a reflux ratio of 10 (a wild1 guess initially, confirmed because the column worked). -

The Deep Hot Biosphere: the Myth of Fossil Fuels Free
FREE THE DEEP HOT BIOSPHERE: THE MYTH OF FOSSIL FUELS PDF Thomas Gold,Freeman J. Dyson | 243 pages | 01 Jun 2001 | Springer-Verlag New York Inc. | 9780387952536 | English | New York, NY, United States The Deep Hot Biosphere: The Myth of Fossil Fuels - Thomas Gold - Google книги Post a comment. Total Pageviews. The Deep Hot Biosphere. It concerns of a microbial biosphere that exists at depth, profuse both within the Earth and probably other planets, feed by primordial and abiotic hydrocarbonsmainly methane and chemical energy. Deep levels in the Earth would be an environment that could form catalytic and autocatalytic molecules that contribute for the emergence of life. After, this deep primeval life developed a sophisticated apparatus to conquest the surface of planet - by photosynthesis - using energy from sunlight to dissociated water and carbon dioxide to produce carbohydrates, permitting autotrophy. The main reason consists in understanding the origin of hydrocarbons whose base is in Deep-Earth Gas Theory. Thomas Gold postulated that hydrocarbons such as oil, natural gas and black coal are primary compounds, i. He stated that hydrocarbons are surelly abiotic compounds and very common on Earth and the universe. In summary his ideas can thus be defined as its own quote:. The deep, hot biosphere. Gold, T. Sci, USA. Vol 89, pp. There are strong indications that microbial life is widespread at depth in the crust of the Earth, just as such life has been identified in numerous ocean vents. This life is not dependent on The Deep Hot Biosphere: The Myth of Fossil Fuels energy and photosynthesis for its primary energy supply, and it is essentially independent of the surface circumstances. -

In the United States District Court for the Northern District of Illinois
IN THE UNITED STATES DISTRICT COURT FOR THE NORTHERN DISTRICT OF ILLINOIS UNITED STATES OF AMERICA, ) STATE OF ILLINOIS, ) STATE OF LOUISIANA, and the ) STATE OF MONTANA ) ) Plaintiffs, ) ) v. ) ) EXXON MOBIL CORPORATION and ) No. EXXONMOBIL OIL CORPORATION ) ) Defendants. ) ) CONSENT DECREE TABLE OF CONTENTS I. JURISDICTION AND VENUE ............................................6 II. APPLICABILITY AND BINDING EFFECT ..................................7 III. OBJECTIVES ..........................................................9 IV. DEFINITIONS ..........................................................9 V. AFFIRMATIVE RELIEF ................................................17 A. Fluid Catalytic Cracking Units ......................................17 B. NOx Emissions Reductions from the FCCUs ...........................18 C. SO2 Emissions Reductions from the FCCUs ............................28 D. Particulate Matter Emissions Reductions from the FCCUs .................39 E. Carbon Monoxide Emissions Reductions from the FCCUs ................41 F. NSPS Applicability to the FCCU Catalyst Regenerators ..................44 G. NOx Emissions Reductions from Combustion Units .....................49 H. SO2 Emissions Reductions from and NSPS Applicability of Heaters, Boilers and Other Fuel Gas Combustion Devices ..............................56 I. Sulfur Recovery Plant Operations ....................................58 J. Flaring Devices ..................................................67 K. Control of Acid Gas Flaring and Tail Gas Incidents ......................71 -

How Did They Finish? CAN ONE MEGASITE HAVE EVERYTHING YOU NEED?
The Best of Aerospace in the American South WWW.RAND LER E P O R T . C OM WWW. SB- D . COM WWW. S O UTHE R N A UTO C O RRI D O R . COM Economic Development in the World’s Third Largest Economy SPRING 2018 2018 SB D100 5 Permit No. 21 No. Permit & A V , g Lynchbur PAID ge Posta .S. U D PRSRT ST PRSRT How did they finish? CAN ONE MEGASITE HAVE EVERYTHING YOU NEED? In the Golden Triangle, we’ve got it all—and we give it all. Yes is what we always say here—for whatever you need to succeed. Abundant electricity and water? Yes! Highways, rail, waterway, airport? Yes, yes, yes, and yes! Quality workforce and training? Hell, yes! Our 1,144-acre Infinity Megasite is just one of our feature-filled, ready-to-roll locations. But what’ll really blow you away is our can-do, will-do, do-it-up attitude. Talk to us. Tell us what you need. And expect the answer that makes this region—and your future—golden: YES. www.gtrSaysYes.com or call 662.328.8369 Joe Max Higgins, Jr., CEO GOLDEN TRIANGLE DEVELOPMENT LINK All of These Companies Have Two Things in Common ... 2.5 million square feet 2 million square feet 2.1 million square feet 2 million square feet 1 million square feet 500,000 square feet Alliance Consulting Engineers, Inc., Nationally Recognized, Recognized as the Locally Focused. Leading Economic Development Engineering Firm in South Carolina by They Chose South Carolina. They Chose Alliance Consulting Engineers, Inc. -
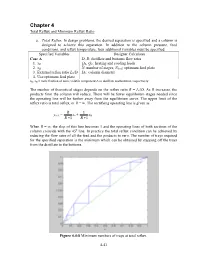
Minimum Reflux Ratio
Chapter 4 Total Reflux and Minimum Reflux Ratio a. Total Reflux . In design problems, the desired separation is specified and a column is designed to achieve this separation. In addition to the column pressure, feed conditions, and reflux temperature, four additional variables must be specified. Specified Variables Designer Calculates Case A D, B: distillate and bottoms flow rates 1. xD QR, QC: heating and cooling loads 2. xB N: number of stages, Nfeed : optimum feed plate 3. External reflux ratio L0/D DC: column diameter 4. Use optimum feed plate xD, xB = mole fraction of more volatile component A in distillate and bottoms, respectively The number of theoretical stages depends on the reflux ratio R = L0/D. As R increases, the products from the column will reduce. There will be fewer equilibrium stages needed since the operating line will be further away from the equilibrium curve. The upper limit of the reflux ratio is total reflux, or R = ∞. The rectifying operating line is given as R 1 yn+1 = xn + xD R +1 R +1 When R = ∞, the slop of this line becomes 1 and the operating lines of both sections of the column coincide with the 45 o line. In practice the total reflux condition can be achieved by reducing the flow rates of all the feed and the products to zero. The number of trays required for the specified separation is the minimum which can be obtained by stepping off the trays from the distillate to the bottoms. Figure 4.4-8 Minimum numbers of trays at total reflux. -

Exxonmobil Oil Spill Response Field Manual
Oil Spill Response Field Manual Metric Conversion Factors Volume Flow Rate 1 cm3 = 1 ml = .001 L 1 L/min = 0.0167 L/sec 1 L = 1 dm3 = .001 m3 = 60 L/hr = 1440 L/day 1 L = 0.264 Gallon Liquid US 1 L/min = 0.06 m3/hr 1 m3 = 6.29 API bbl 1 L/min = 0.265 gpm (US) 1 L/min = 9.05 API bbl/day Length Velocity 1 m = 10–3 km = 102 cm = 103 mm = 106µ 1 cm/sec = 10–2 m/sec = 36 m/hr 1 cm = 0.3937 in = 0.036 km/hr 1 m = 3.2808 ft = 39.37 in 1 m/sec = 1.94 knots (US) 1 m = 0.5468 fathom 1 km/hr = 0.54 knots (US) 1 km = 0.62 mile = 3273 ft 1 km/hr = 0.621 mph (US) 1 km = 0.54 nautical mile (NM) Area Mass/Weight 1 hectare = 10,000 m2 = 0.01 km2 1g = 10–3 kg = 103 mg 1 m2 = 10.76 ft2 = 1.196 yd2 1 metric tonne = 1000 kg 1 hectare = 2.471 acres = 0.00386 sq mile 1 kg = 2.21 lbs = 0.0685 slug Surface Tension Force 1 kg-f/m = 9.807 N/m = 9807 dyne/cm 1 newton (N) = 105 dyne 1 kg-f/m = 0.672 lbs/ft = 5.61 lb/in 1 newton = 0.102 kg-f 1 N/m = 0.0685 lb/ft 1 newton = 0.2248 lb 1 N/mm = 5.64 lb/in 1 newton = 7.233 pdl Pressure Application Rates 1 N/m2 = 0.102 kg-f/m2 = 1 pascal (Pa) 1 L/m2 = thickness in mm 1 bar = 106 dyne/cm2 = 0.1MPa 1 L/hectare = 0.1 m3/km2 1 mm Hg = 133 Pa 1 L/hectare = 0.1068 gal/acre 1 Pa = 1.450 x 10–4 psi 1 tonne/hectare = 2.5 bbl/acre 1 kg-f/m2 = 0.0206 lb/ft2 1 MPa = 9.869 atm Miscellaneous 1 tonne of oil = 1000 L = 1 m3 = 264.2 gal storage volume for boom, volume/length: ft3/ft x 0.093 = m3/m mg/L = parts per million (ppm) = % x 10–2 x 106 = ppm ice density = 0.8 g/cm3 = 800 kg/m3 viscosity in centipoise (cp) = viscosity in centistokes (cSt) x density temperature centigrade = (temperature Fahrenheit – 32) x 0.555 Oil Spill Response Field Manual Revised 2008 Copyright © 2008, ExxonMobil Research and Engineering Company, All Rights Reserved, Printed in USA. -

Alexander II and Gorbachev: the Doomed Reformers of Russia
University of Vermont UVM ScholarWorks UVM Honors College Senior Theses Undergraduate Theses 2017 Alexander II and Gorbachev: The Doomed Reformers of Russia Kate V. Lipman The University of Vermont Follow this and additional works at: https://scholarworks.uvm.edu/hcoltheses Recommended Citation Lipman, Kate V., "Alexander II and Gorbachev: The Doomed Reformers of Russia" (2017). UVM Honors College Senior Theses. 158. https://scholarworks.uvm.edu/hcoltheses/158 This Honors College Thesis is brought to you for free and open access by the Undergraduate Theses at UVM ScholarWorks. It has been accepted for inclusion in UVM Honors College Senior Theses by an authorized administrator of UVM ScholarWorks. For more information, please contact [email protected]. 1 Alexander II and Gorbachev: The Doomed Reformers of Russia Kate V. Lipman Thesis Submitted for College Honors in History Advisor: Denise J. Youngblood The University of Vermont May 2017 2 Note on Sources: There is, surprisingly, little available in English on the reign of Alexander II, therefore I rely primarily on the works of W.E. Mosse, N.G.O Pereira, and the collection of works on Russia’s reform period edited by Ben Eklof. W.E. Mosse’s book Alexander II and the Modernization of Russia, a secondary source, provides in-depth descriptions of the problems facing Alexander II when he came into power, the process of his reforms, descriptions of the reforms, and some of the consequences of those reforms including his assassination in 1881. Similarly, N.G.O Pereira’s Tsar-Liberator: Alexander II of Russia 1818-1881 is a secondary source, describing some of his reforms, but focuses most specifically on the process of the emancipation of the serfs, the emancipation itself and the impact it had on the population.