Structure Elucidation and Isotopic Labeling Experiments Provide Insights Into the Biosynthesis of Three Phytochemicals: Alkamides, Avenacin, And
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biosynthesis of New Alpha-Bisabolol Derivatives Through a Synthetic Biology Approach Arthur Sarrade-Loucheur
Biosynthesis of new alpha-bisabolol derivatives through a synthetic biology approach Arthur Sarrade-Loucheur To cite this version: Arthur Sarrade-Loucheur. Biosynthesis of new alpha-bisabolol derivatives through a synthetic biology approach. Biochemistry, Molecular Biology. INSA de Toulouse, 2020. English. NNT : 2020ISAT0003. tel-02976811 HAL Id: tel-02976811 https://tel.archives-ouvertes.fr/tel-02976811 Submitted on 23 Oct 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE En vue de l’obtention du DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE Délivré par l'Institut National des Sciences Appliquées de Toulouse Présentée et soutenue par Arthur SARRADE-LOUCHEUR Le 30 juin 2020 Biosynthèse de nouveaux dérivés de l'α-bisabolol par une approche de biologie synthèse Ecole doctorale : SEVAB - Sciences Ecologiques, Vétérinaires, Agronomiques et Bioingenieries Spécialité : Ingénieries microbienne et enzymatique Unité de recherche : TBI - Toulouse Biotechnology Institute, Bio & Chemical Engineering Thèse dirigée par Gilles TRUAN et Magali REMAUD-SIMEON Jury -

Isolation, Identification and Characterization of Allelochemicals/Natural Products
Isolation, Identification and Characterization of Allelochemicals/Natural Products Isolation, Identification and Characterization of Allelochemicals/Natural Products Editors DIEGO A. SAMPIETRO Instituto de Estudios Vegetales “Dr. A. R. Sampietro” Universidad Nacional de Tucumán, Tucumán Argentina CESAR A. N. CATALAN Instituto de Química Orgánica Universidad Nacional de Tucumán, Tucumán Argentina MARTA A. VATTUONE Instituto de Estudios Vegetales “Dr. A. R. Sampietro” Universidad Nacional de Tucumán, Tucumán Argentina Series Editor S. S. NARWAL Haryana Agricultural University Hisar, India Science Publishers Enfield (NH) Jersey Plymouth Science Publishers www.scipub.net 234 May Street Post Office Box 699 Enfield, New Hampshire 03748 United States of America General enquiries : [email protected] Editorial enquiries : [email protected] Sales enquiries : [email protected] Published by Science Publishers, Enfield, NH, USA An imprint of Edenbridge Ltd., British Channel Islands Printed in India © 2009 reserved ISBN: 978-1-57808-577-4 Library of Congress Cataloging-in-Publication Data Isolation, identification and characterization of allelo- chemicals/natural products/editors, Diego A. Sampietro, Cesar A. N. Catalan, Marta A. Vattuone. p. cm. Includes bibliographical references and index. ISBN 978-1-57808-577-4 (hardcover) 1. Allelochemicals. 2. Natural products. I. Sampietro, Diego A. II. Catalan, Cesar A. N. III. Vattuone, Marta A. QK898.A43I86 2009 571.9’2--dc22 2008048397 All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying or otherwise, without the prior permission of the publisher, in writing. The exception to this is when a reasonable part of the text is quoted for purpose of book review, abstracting etc. -

The Biosynthesis of Sesquiterpene Lactones in Chicory (Cichorium Intybus L.) Roots Promotor Prof
The Biosynthesis of Sesquiterpene Lactones in Chicory (Cichorium intybus L.) Roots Promotor Prof. dr. Æ. de Groot, hoogleraar in de bio-organische chemie, Wageningen Universiteit Co-promotoren Dr. M.C.R. Franssen, universitair hoofddocent, Laboratorium voor Organische Chemie, Wageningen Universiteit Dr. ir. H.J. Bouwmeester, senior onderzoeker, Business Unit Celcybernetica, Plant Research International Promotiecommissie Prof. dr. J. Gershenzon (Max Planck Institute for Chemical Ecology, Germany) Prof. dr. L.H.W. van der Plas (Wageningen Universiteit) Prof. dr. ir. I.M.C.M. Rietjens (Wageningen Universiteit) Prof. dr. E.J.R. Sudhölter (Wageningen Universiteit) Jan-Willem de Kraker The Biosynthesis of Sesquiterpene Lactones in Chicory (Cichorium intybus L.) Roots Proefschrift ter verkrijging van de graad van doctor op gezag van de rector magnificus van Wageningen Universiteit prof. dr. ir. L. Speelman in het openbaar te verdedigen op woensdag 9 januari 2002 des namiddags te vier uur in de Aula de Kraker, J.-W. The Biosynthesis of Sesquiterpene Lactones in Chicory (Cichorium intybus L.) Roots Thesis Wageningen University – with summaries in English and Dutch ISBN 90-5808-531-7 Voorwoord — Preface De afgelopen vijf jaar was het voor mij witlof, dag in dag uit. Waarschijnlijk is dit ook de reden dat ik al ruim 3 jaar geen witlof meer kan ruiken, laat staan eet. Als je dan ongeacht waar je in Europa bent overal in de wegberm uitsluitend blauwe bloemen ziet opduiken, is het wel goed en verstandig er eens een punt achter te zetten. Hoewel het eind van dit promotie- onderzoek door al het ‘wachtgeldgedoe’ en bijbehorende ‘dode mussen’ wel erg abrupt en weinig chique kwam, kan ik me nu met goed gevoel weer eens op wat anders storten. -

(12) United States Patent (10) Patent No.: US 7,214.507 B2 Bouwmeester Et Al
US007214507B2 (12) United States Patent (10) Patent No.: US 7,214.507 B2 BOuWmeester et al. (45) Date of Patent: May 8, 2007 (54) PLANT ENZYMES FOR BIOCONVERSION (56) References Cited (75) Inventors: Hendrik Jan Bouwmeester, Renkum U.S. PATENT DOCUMENTS (NL); Jan-Willem de Kraker, Jena 6,200,786 B1* 3/2001 Huang et al. ............... 435,132 (DE); Marloes Schurink, Ede (NL); 6,451,576 B1* 9/2002 Croteau et al. ............. 435/232 Raoul John Bino, Wageningen (NL); Aede de Groot, Wageningen (NL); OTHER PUBLICATIONS Maurice Charles R. Franssen, Dedeyan B et al. 2000. Appl Env Microbiol 66: 925-929.* Wageningen (NL) Yoder OC et al. 2001. Curr Opin Plant Biol 4: 315-321.* Nielsen KAetal. 2005. Cytochrome P450s in plants. In Cytochrome (73) Assignee: Plant Research International B.V., P450: structure, mechanism, and biochemistry, 3' ed., Ortiz de Wageningen (NL) Montellano, ed., p. 553.* Bohlmann, Jorg, et al., “Plant terpenoid synthases: Molecular (*) Notice: Subject to any disclaimer, the term of this biology and phylogenetic analysis”. Proc. Natl. Acad. Sci. 1998, patent is extended or adjusted under 35 95:4126-4133. de Kraker, Jan-Willem, et al., “Biosynthesis of Germacrene A U.S.C. 154(b) by 0 days. Carboxylic Acid in Chicory Roots. Demonstration of a Cytochrome P450 (+)-Germacrene A Hydroxylase and NADP-Dependent (21) Appl. No.: 10/489,762 Sesquiterpenoid Dehydrogenase(s) Involved in Sesquiterpene Lactone Biosynthesis”. Plant Physiology 2001, 125:1930-1940. (22) PCT Filed: Sep. 17, 2002 de Kraker, Jan-Willem, et al., “Germacrenes from fresh costus roots”, Phytochemistry 2001, 58:481-487. (86). PCT No.: PCT/NLO2/OO591 de Kraker, Jan-Willem, et al., “(+)-Germacrene A Biosynthesis'. -

Costunolide—A Bioactive Sesquiterpene Lactone with Diverse Therapeutic Potential
Review Costunolide—A Bioactive Sesquiterpene Lactone with Diverse Therapeutic Potential Dae Yong Kim 1 and Bu Young Choi 2,* 1 Department of Biology Education, Seowon University, Cheongju, Chungbuk 361-742, So Korea; [email protected] 2 Department of Pharmaceutical Science & Engineering, Seowon University, Cheongju, Chungbuk 361-742, South Korea * Correspondance: [email protected]; Tel.: +82-043-299-8411 Received: 23 April 2019; Accepted: 12 June 2019; Published: 14 June 2019 Abstract: Sesquiterpene lactones constitute a major class of bioactive natural products. One of the naturally occurring sesquiterpene lactones is costunolide, which has been extensively investigated for a wide range of biological activities. Multiple lines of preclinical studies have reported that the compound possesses antioxidative, anti-inflammatory, antiallergic, bone remodeling, neuroprotective, hair growth promoting, anticancer, and antidiabetic properties. Many of these bioactivities are supported by mechanistic details, such as the modulation of various intracellular signaling pathways involved in precipitating tissue inflammation, tumor growth and progression, bone loss, and neurodegeneration. The key molecular targets of costunolide include, but are not limited to, intracellular kinases, such as mitogen-activated protein kinases, Akt kinase, telomerase, cyclins and cyclin-dependent kinases, and redox-regulated transcription factors, such as nuclear factor-kappaB, signal transducer and activator of transcription, activator protein-1. The compound also diminished the production and/expression of proinflammatory mediators, such as cyclooxygenase-2, inducible nitric oxide synthase, nitric oxide, prostaglandins, and cytokines. This review provides an overview of the therapeutic potential of costunolide in the management of various diseases and their underlying mechanisms. Keywords: costunolide; antioxidants; anti-inflammatory; anti-allergic; bone regenerating; neuroprotective; antimicrobial; hair growth promoting; anticancer; antidiabetic properties 1. -

Isolation and Characterization of Two Germacrene a Synthase Cdna Clones from Chicory1
Isolation and Characterization of Two Germacrene A Synthase cDNA Clones from Chicory1 Harro J. Bouwmeester*, Jan Kodde, Francel W.A. Verstappen, Iris G. Altug, Jan-Willem de Kraker, and T. Eelco Wallaart2 Plant Research International, Business Unit Cell Cybernetics, P.O. Box 16, 6700 AA Wageningen, The Netherlands (H.J.B., J.K., F.W.A.V.); Department of Organic Chemistry, Hamburg University, D–20146 Hamburg, Germany (I.G.A.); Department of Organic Chemistry, Wageningen Agricultural University, Dreijenplein 8, 6703 HB Wageningen, The Netherlands (J.-W.d.K.); and University Centre for Pharmacy, Department of Pharmaceutical Biology, University of Groningen, Antonius Deusinglaan 1, 9713 AV Groningen, The Netherlands (T.E.W.) Chicory (Cichorium intybus) sesquiterpene lactones were recently shown to be derived from a common sesquiterpene intermediate, (ϩ)-germacrene A. Germacrene A is of interest because of its key role in sesquiterpene lactone biosynthesis and because it is an enzyme-bound intermediate in the biosynthesis of a number of phytoalexins. Using polymerase chain reaction with degenerate primers, we have isolated two sesquiterpene synthases from chicory that exhibited 72% amino acid identity. Heterologous expression of the genes in Escherichia coli has shown that they both catalyze exclusively the formation of (ϩ)-germacrene A, making this the first report, to our knowledge, on the isolation of (ϩ)-germacrene A synthase (GAS)-encoding genes. Northern analysis demonstrated that both genes were expressed in all chicory tissues tested albeit at varying levels. Protein isolation and partial purification from chicory heads demonstrated the presence of two GAS proteins. On MonoQ, these proteins co-eluted with the two heterologously produced proteins. -

Biochemical and Functional Characterization of Induced Terpene Formation in Arabidopsis Roots
Biochemical and Functional Characterization of Induced Terpene Formation in Arabidopsis Roots Reza Sohrabi Dissertation submitted to the faculty of the Virginia Polytechnic Institute and State University in partial fulfillment of the requirements for the degree of Doctor of Philosophy In Biological Sciences Dorothea Tholl, Committee Chair Glenda Gillaspy Khidir Hilu John Jelesko July 23rd 2013 Blacksburg Virginia Keywords: Cytochrome P450, plant volatiles, specialized metabolism, root chemical defense, Arabidopsis iii Biochemical and Functional Characterization of Induced Terpene Formation in Arabidopsis Roots Reza Sohrabi ABSTRACT Plants have evolved a variety of constitutive and induced chemical defense mechanisms against biotic stress. Emission of volatile compounds from plants facilitates interactions with both beneficial and pathogenic organisms. However, knowledge of the chemical defense in roots is still limited. In this study, we have examined the root-specific biosynthesis and function of volatile terpenes in the model plant Arabidopsis. When infected with the root rot pathogen Pythium irregulare, Arabidopsis roots release the acyclic C11-homoterpene (E)-4,8- dimethylnona-1,3,7-triene (DMNT), which is a common constituent of volatile blends emitted from insect-damaged foliage. We have identified a single cytochrome P450 monooxygenase of the CYP705 family that catalyzes a root-specific oxidative degradation of the C30-triterpene precursor arabidiol thereby causing the release of DMNT and a C19-degradation product named arabidonol. We found that DMNT shows inhibitory effects on P. irregulare mycelium growth and oospore germination in vitro, and that DMNT biosynthetic mutant plants were more susceptible to P. irregulare infection. We provide evidence based on genome synteny and phylogenetic analysis that the arabidiol biosynthetic gene cluster containing the arabidiol synthase (ABDS) and CYP705A1 genes possibly emerged via local gene duplication followed by iv de novo neofunctionalization. -

Elucidation of the Sesquiterpene Lactone Biosynthetic Pathway in Feverfew (Tanacetum Parthenium)
Elucidation of the sesquiterpene lactone biosynthetic pathway in feverfew (Tanacetum parthenium) Qing Liu Thesis committee Promotor Prof. dr. ir. H.J. Bouwmeester Professor of Plant Physiology Wageningen University Co-promotor Dr. S.V.D. Krol Associate professor, Laboratory of Plant Physiology Wageningen University Other members Prof. dr. A. Ferrer, Centre for Research in Agricultural Genomics, Bellaterra, Spain Dr. M.C.R. Franssen, Wageningen University Prof. dr. M.E. Schranz, Wageningen University Dhr. dr. ir. R.C. Schuurink, University of Amsterdam This research was conducted under the auspices of the Graduate School of Experimental Plant Sciences. Elucidation of the sesquiterpene lactone biosynthetic pathway in feverfew (Tanacetum parthenium) Qing Liu Thesis submitted in fulfilment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Monday 2nd December 2013 at 1:30 p.m. in the Aula. Qing Liu Elucidation of the sesquiterpene lactone biosynthetic pathway in feverfew (Tanacetum parthenium) 134 pages PhD thesis, Wageningen University, Wageningen, the Netherlands (2013) With summaries in English and Dutch ISBN 978-94-6173-756-4 Contents Chapter 1 General introduction 7 Chapter 2 Biosynthesis and localization of parthenolide in glan- dular trichomes of feverfew (Tanacetum parthenium L. Schulz Bip.) 19 Chapter 3 Reconstitution of the costunolide biosynthetic -
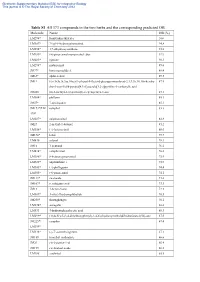
Table S1 All 373 Compounds in the Two Herbs and the Corresponding
Electronic Supplementary Material (ESI) for Integrative Biology This journal is © The Royal Society of Chemistry 2012 Table S1 All 373 compounds in the two herbs and the corresponding predicted OB Molecule Name OB (%) LM294* forsythidmethylester 100 LM387* 7'-epi-8-hydroxypinoresinol 94.8 LM394* 1,7-dihydroxyxanthone 93.8 LM289* (+)-pinoresinol monomethyl ether 92.9 LM285* egenine 90.3 LM298* matairesinol 89.6 JM77* loniceracetalide A 89.4 JM63* alpha-cedrol 89.3 JM1* (-)-(3r,8s,9r,9as,10as)-9-ethenyl-8-(beta-d-glucopyranosyloxy)-2,3,9,9a,10,10a-hexahy 87.5 dro-5-oxo-5h,8h-pyrano[4,3-d]oxazolo[3,2-a]pyridine-3-carboxylic acid JM200 (z)-3-methyl-2-(2-pentenyl)-2-cyclopenten-1-one 87.1 LM304* phillyrin 85.1 JM57* 7-epi-loganin 85.1 JM173*/LM camphol 83.3 398* LM287* epipinoresinol 82.8 JM27 2-methyl-1-butanol 81.2 LM356* (+)-lariciresinol 80.0 JM170* ledol 79.7 LM430 safynol 78.3 JM16 1-pentanol 76.2 LM414* campherenol 76.0 LM386* 8-hydroxypinoresinol 75.9 LM428* onjixanthone i 75.9 LM345* (+)-phillygenin 74.4 LM305* (+)-pinoresinol 74.1 JM113* sweroside 73.6 JM107* secologanic acid 73.3 JM13 1-hexen-3-one 72.3 LM309* 3-ethyl-7hydroxyphthalide 70.5 JM250* shuangkangsu 70.1 LM274* astragalin 68.6 LM331 4-hydroxyphenylacetic acid 68.3 LM299* (3r,4s,5r)-5-(3,4-dimethoxyphenyl)-3,4-bis(hydroxymethyl)dihydrofuran-2(3h)-one 67.5 JM225*/ camphor 67.4 LM399* LM338* (-)-7’-o-methylegenine 67.1 JM189 trimethyl isothiazole 66.6 JM29 cis-2-penten-1-ol 66.4 JM199 cis-linalool oxide 66.2 LM288 erythritol 65.8 Electronic Supplementary Material (ESI) for -

Cytochrome P450S and Uses Thereof Joe Chappell University of Kentucky, [email protected]
University of Kentucky UKnowledge Plant and Soil Sciences Faculty Patents Plant and Soil Sciences 9-11-2012 Cytochrome P450S and Uses Thereof Joe Chappell University of Kentucky, [email protected] Lyle F. Ralston Right click to open a feedback form in a new tab to let us know how this document benefits oy u. Follow this and additional works at: https://uknowledge.uky.edu/pss_patents Part of the Plant Sciences Commons Recommended Citation Chappell, Joe and Ralston, Lyle F., "Cytochrome P450S and Uses Thereof" (2012). Plant and Soil Sciences Faculty Patents. 9. https://uknowledge.uky.edu/pss_patents/9 This Patent is brought to you for free and open access by the Plant and Soil Sciences at UKnowledge. It has been accepted for inclusion in Plant and Soil Sciences Faculty Patents by an authorized administrator of UKnowledge. For more information, please contact [email protected]. US008263362B2 (12) United States Patent (10) Patent N0.: US 8,263,362 B2 Chappell et a]. (45) Date of Patent: *Sep. 11, 2012 (54) CYTOCHROME P450S AND USES THEREOF 2003/0166255 A1 9/2003 Chappell .................. .. 435/2523 2004/0078840 A1 4/2004 Chappell 6151. .. .. 800/278 2006/0218661 A1 9/2006 Chappell 6151. .. .. 800/278 (75) Inventors: Joseph Chappell, Lexington, KY (US); 2007/0231861 A1 10/2007 Millis et a1. 435/69.l Lyle F. Ralston, Kirkwood, MO (US) 2007/0238157 A1 10/2007 Millis et a1. .. 435/166 2007/0238159 A1 10/2007 Millis et a1. 435/25233 (73) Assignee: University of Kentucky Research 2007/0238160 A1 10/2007 Millis et a1. 435/25233 Foundation, Lexington, KY (US) 2007/0254354 A1 11/2007 Millis et a1. -

Asteraceae) Family
University of Calgary PRISM: University of Calgary's Digital Repository Graduate Studies The Vault: Electronic Theses and Dissertations 2013-05-06 Biochemical and evolutionary studies of sesquiterpene lactone metabolism in the sunflower (Asteraceae) family Nguyen, Trinh Don Nguyen, T. D. (2013). Biochemical and evolutionary studies of sesquiterpene lactone metabolism in the sunflower (Asteraceae) family (Unpublished doctoral thesis). University of Calgary, Calgary, AB. doi:10.11575/PRISM/25121 http://hdl.handle.net/11023/702 doctoral thesis University of Calgary graduate students retain copyright ownership and moral rights for their thesis. You may use this material in any way that is permitted by the Copyright Act or through licensing that has been assigned to the document. For uses that are not allowable under copyright legislation or licensing, you are required to seek permission. Downloaded from PRISM: https://prism.ucalgary.ca UNIVERSITY OF CALGARY Biochemical and evolutionary studies of sesquiterpene lactone metabolism in the sunflower (Asteraceae) family by Trinh Don Nguyen A THESIS SUBMITTED TO THE FACULTY OF GRADUATE STUDIES IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY DEPARTMENT OF BIOLOGICAL SCIENCES CALGARY, ALBERTA APRIL, 2013 © Trinh Don Nguyen 2013 Abstract Plants have evolved the capacity to synthesize a myriad of specialized metabolites which enhance their fitness in specific living conditions. These compounds are also widely utilized for human purposes. Elucidating the enzymes in plant specialized metabolism has been one of the main forces driving plant biochemistry. The more intriguing question, however, is how these enzymes evolved to acquire their existent functions. The Asteraceae, the largest flowering plant family, is well-known for its enormously diverse and lineage-characteristic contents of sesquiterpene lactones (STLs). -

Exploring the Biochemical and Evolutionary Diversity of Terpene Biosynthetic Enzymes in Plants
University of Kentucky UKnowledge University of Kentucky Doctoral Dissertations Graduate School 2008 EXPLORING THE BIOCHEMICAL AND EVOLUTIONARY DIVERSITY OF TERPENE BIOSYNTHETIC ENZYMES IN PLANTS Sungbeom Lee University of Kentucky, [email protected] Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Recommended Citation Lee, Sungbeom, "EXPLORING THE BIOCHEMICAL AND EVOLUTIONARY DIVERSITY OF TERPENE BIOSYNTHETIC ENZYMES IN PLANTS" (2008). University of Kentucky Doctoral Dissertations. 587. https://uknowledge.uky.edu/gradschool_diss/587 This Dissertation is brought to you for free and open access by the Graduate School at UKnowledge. It has been accepted for inclusion in University of Kentucky Doctoral Dissertations by an authorized administrator of UKnowledge. For more information, please contact [email protected]. ABSTRACT OF DISSERTATION Sungbeom Lee The Graduate School University of Kentucky 2008 EXPLORING THE BIOCHEMICAL AND EVOLUTIONARY DIVERSITY OF TERPENE BIOSYNTHETIC ENZYMES IN PLANTS ABSTRACT OF DISSERTATION A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Plant and Soil Sciences at the University of Kentucky By Sungbeom Lee Lexington, Kentucky Director: Dr. Joseph Chappell, Professor Lexington, Kentucky 2008 Copyright © Sungbeom Lee 2008 ABSTRACT OF DISSERTATION EXPLORING THE BIOCHEMICAL AND EVOLUTIONARY DIVERSITY OF TERPENE BIOSYNTHETIC ENZYMES IN PLANTS Southern Magnolia (Magnolia grandiflora) is a primitive tree species that has attracted attention because of its horticultural distinctiveness, the wealth of natural products associated with it, and its evolutionary position as a basal angiosperm. Terpenoid constituents were determined from Magnolia leaves and flowers. Magnolia leaves constitutively produced two major terpenoids, β-cubebene and germacrene A.