Coagulation and Clot Breakdown
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

We Get Biased on Things That Make Sense
DIALOGUE AND DISCUSSION We Get Biased on Things That Make Sense MERIH T. TESFAZGHI When I was teaching third year medical students, and bench- reaction might be characterized by a marked left shift lecturing clinical laboratory science students, I emphasized which may display a range of immature cells that are also the importance of clinical details and the history of patients seen in CML. Severe left shift especially in the absence of in laboratory test processing, especially in the evaluation of evident infection (or other underlying diseases) may peripheral blood films. During those times, one of my strongly suggest direct bone marrow involvement. In students stated, “but including details of patients might such scenario, clinical details of patients such as absence somehow affect the decision of the reviewer, and may lead to or presence of enlargement of extramedullar organs is Downloaded from a bias.” I responded that we are biased on things that make highly sought. sense. This means that we decide based on the evidence we see from a microscopy evaluation in the light of the clinical Ø The coexistence of multiple conditions in a given detail and history. How do clinical details and history help us patient. One condition may mask the presence of the reach decisions? other condition. For example, the coexistence of megaloblastic anemia with iron deficiency anemia. http://hwmaint.clsjournal.ascls.org/ If specimens are the “in vitro ambassadors” of patients to the Usually, megaloblastic anemia is characterized by laboratory, then properly completed request forms are their increased size of red blood cells (MCV), but when “credentials”. -
Hematology Unit Lab 1 Review Material
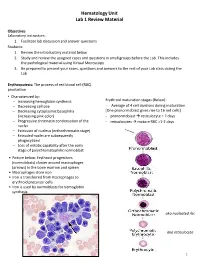
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Copyrighted Material
CHAPTER 1 The Blood Film and Count Blood Blood is a life‐sustaining fluid that circulates through the heart and blood vessels. It carries oxygen and nutrients to the tissues and waste products to the lungs, liver and kidneys, where they can be removed from the body. Usually when blood is removed from the body it forms a solid blood clot. However, if clotting is prevented by mixing with an anticoagulant, the blood separates, under the influence of gravity, into three layers (Fig. 1.1). The bottom layer is deep red in colour and is composed of red cells. The top layer is clear and pale yellow. It is called plasma and is composed of various salts and proteins dissolved in water. In between is a narrow layer called the buffy coat because of its buff or yellowish white colour. The buffy coat is composed mainly of cells of a variety of types, collectively known as white cells. In addition there are small cellular fragments, called platelets, which have a role in blood clotting. The bloodCOPYRIGHTED film MATERIAL Although we can judge the proportions of red cells and white cells in a tube of sedimented blood, we get far more information if the blood is carefully mixed and a thin layer is spread on a glass A Beginner’s Guide to Blood Cells, Third Edition. Barbara J. Bain. © 2017 John Wiley & Sons Ltd. Published 2017 by John Wiley & Sons Ltd. 1 0003049575.indd 1 2/24/2017 9:50:31 AM 2 A Beginner’s Guide to Blood Cells Plasma Buffy coat Red cells Fig. -

Blood Film Preparation and Staining Procedures
INTERPRETATION OF THE PERIPHERAL 00. ם BLOOD FILM 0272–2712/02 $15.00 BLOOD FILM PREPARATION AND STAINING PROCEDURES Berend Houwen, MD, PhD The blood film is one of the world’s most widely and frequently used tests and has undergone remarkably few changes since its introduction as a clinical diagnostic tool in the late 1800s. The origins of stained blood film microscopy are somewhat obscure, and a clear reference for the ‘‘first person or persons’’ to describe its use as a clinical laboratory procedure cannot be established with certainty. Antonie van Leeuwenhoek, in the seventeenth century, was the first to describe blood cells, using whole blood preparations and not blood films for his observations on blood corpuscles. In fact, he would not have been able to use blood film–based microscopy because it requires considerably more sophisti- cated optics than were available in his day. A second technologic innovation enabling blood film microscopy was the introduction of aniline dyes in the second half of the nineteenth century. This made it possible to study individual blood cells by light microscopy after a small amount of blood had been placed and smeared onto glass slides, dried, fixed, and then stained. Paul Ehrlich introduced eosin as the first of these dyes for staining blood films in 1856, followed by hematoxylin in 1865, and later by the metachromatic Romanowsky dyes. A few refinements have since been made, consisting mainly of improve- ments in dye quality, staining procedures, automation of slide preparation, and staining, but the basic elements of blood film preparation and analysis have not changed for over a century. -

Platelet Function Disorders
TREATMENT OF HEMOPHILIA APRIL 2008 • NO 19 PLATELET FUNCTION DISORDERS Second Edition Anjali A. Sharathkumar Amy Shapiro Indiana Hemophilia and Thrombosis Center Indianapolis, U.S.A. Published by the World Federation of Hemophilia (WFH), 1999; revised 2008. © World Federation of Hemophilia, 2008 The WFH encourages redistribution of its publications for educational purposes by not-for-profit hemophilia organizations. In order to obtain permission to reprint, redistribute, or translate this publication, please contact the Communications Department at the address below. This publication is accessible from the World Federation of Hemophilia’s website at www.wfh.org. Additional copies are also available from the WFH at: World Federation of Hemophilia 1425 René Lévesque Boulevard West, Suite 1010 Montréal, Québec H3G 1T7 CANADA Tel. : (514) 875-7944 Fax : (514) 875-8916 E-mail: [email protected] Internet: www.wfh.org The Treatment of Hemophilia series is intended to provide general information on the treatment and management of hemophilia. The World Federation of Hemophilia does not engage in the practice of medicine and under no circumstances recommends particular treatment for specific individuals. Dose schedules and other treatment regimes are continually revised and new side effects recognized. WFH makes no representation, express or implied, that drug doses or other treatment recommendations in this publication are correct. For these reasons it is strongly recommended that individuals seek the advice of a medical adviser and/or consult printed instructions provided by the pharmaceutical company before administering any of the drugs referred to in this monograph. Statements and opinions expressed here do not necessarily represent the opinions, policies, or recommendations of the World Federation of Hemophilia, its Executive Committee, or its staff. -

Complete Blood Count in Primary Care
Complete Blood Count in Primary Care bpac nz better medicine Editorial Team bpacnz Tony Fraser 10 George Street Professor Murray Tilyard PO Box 6032, Dunedin Clinical Advisory Group phone 03 477 5418 Dr Dave Colquhoun Michele Cray free fax 0800 bpac nz Dr Rosemary Ikram www.bpac.org.nz Dr Peter Jensen Dr Cam Kyle Dr Chris Leathart Dr Lynn McBain Associate Professor Jim Reid Dr David Reith Professor Murray Tilyard Programme Development Team Noni Allison Rachael Clarke Rebecca Didham Terry Ehau Peter Ellison Dr Malcolm Kendall-Smith Dr Anne Marie Tangney Dr Trevor Walker Dr Sharyn Willis Dave Woods Report Development Team Justine Broadley Todd Gillies Lana Johnson Web Gordon Smith Design Michael Crawford Management and Administration Kaye Baldwin Tony Fraser Kyla Letman Professor Murray Tilyard Distribution Zane Lindon Lyn Thomlinson Colleen Witchall All information is intended for use by competent health care professionals and should be utilised in conjunction with © May 2008 pertinent clinical data. Contents Key points/purpose 2 Introduction 2 Background ▪ Haematopoiesis - Cell development 3 ▪ Limitations of reference ranges for the CBC 4 ▪ Borderline abnormal results must be interpreted in clinical context 4 ▪ History and clinical examination 4 White Cells ▪ Neutrophils 5 ▪ Lymphocytes 9 ▪ Monocytes 11 ▪ Basophils 12 ▪ Eosinophils 12 ▪ Platelets 13 Haemoglobin and red cell indices ▪ Low haemoglobin 15 ▪ Microcytic anaemia 15 ▪ Normocytic anaemia 16 ▪ Macrocytic anaemia 17 ▪ High haemoglobin 17 ▪ Other red cell indices 18 Summary Table 19 Glossary 20 This resource is a consensus document, developed with haematology and general practice input. We would like to thank: Dr Liam Fernyhough, Haematologist, Canterbury Health Laboratories Dr Chris Leathart, GP, Christchurch Dr Edward Theakston, Haematologist, Diagnostic Medlab Ltd We would like to acknowledge their advice, expertise and valuable feedback on this document. -

Can Hemoglobin-Hematocrit Relationship Be Used to Assess Hydration Status?
OPINI Can Hemoglobin-Hematocrit Relationship Be Used to Assess Hydration Status? Hubertus Hosti Hayuanta Resident in the Department of Clinical Pathology, Faculty of Medicine, University of Indonesia, Jakarta, Indonesia ABSTRACT There is an opinion that if the hematocrit is lower than multiplied hemoglobin (Hct< 3 x Hb), the patient is overhydrated, and if it is higher (Hct> 3 x Hb), the patient is dehydrated. This practice is flawed. Hemoglobin-hematocrit relationship is not affected by a patient’s hydration status, and thus its alteration cannot be used to assess it. The relationship can only be altered if the red blood cells (RBCs) are abnormal, or look altered because of technical factors.Instead of multiplying hemoglobin value and comparing it to the hematocrit, a quicker way to assess is to evaluate the mean corpuscular hemoglobin concentration (MCHC). Clinicians can still predict hydration status by comparing the hematocrit to its baseline value or the laboratory’s reference range, by physical examination, or use other laboratory tests such as urine specifi c gravity and osmolality. Keywords: Hematocrit, hemoglobin, hydration ABSTRAK Ada pendapat bahwa jika nilai hematokrit lebih rendah dari nilai kadar hemoglobin dikalikan tiga maka pasien mengalami kelebihan cairan, dan jika lebih tinggi maka pasien mengalami dehidrasi. Praktik penilaian seperti ini tidak benar. Hubungan hemoglobin-hematokrit tidak dipengaruhi oleh status hidrasi pasien, dan oleh karena itu tidak dapat digunakan untuk menilainya. Hubungan tersebut hanya bisa diubah jika eritrosit tidak normal, atau terlihat seakan-akan berubah karena faktor teknis. Cara yang lebih cepat adalah menilai mean corpuscular hemoglobin concentration (MCHC). Klinisi masih bisa menilai status hidrasi dengan membandingkan hematokrit dengan nilai basalnya atau nilai rujukan laboratorium, dengan pemeriksaan fisik, atau melakukan pemeriksaan laboratorium lain seperti berat jenis urin dan osmolalitas. -

TITLE: Complete Blood Count (CBC) Basics PRESENTER: Sarah Drawz and Michael Linden
TITLE: Complete Blood Count (CBC) Basics PRESENTER: Sarah Drawz and Michael Linden Slide 1: Good afternoon, my name is Dr. Michael Linden. I am the Director of Hematopathology at the University of Minnesota in Minneapolis, MN. With me is Dr. Sarah Drawz, who is a Hematopathology Fellow, also at the University of Minnesota. I would like to introduce Dr. Sarah Drawz, who will be beginning our presentation as part of this AACC Pearl of Laboratory Medicine on “Complete Blood Count (CBC) Basics.” Slide 2: The complete blood count (CBC) is an important initial test in the evaluation of a patient’s hematologic function. The CBC is performed on whole blood, which is composed of two primary components: plasma and cells. The plasma fraction contains mostly water, numerous proteins, electrolytes, and clotting factors. The cellular component of blood is comprised of the three major categories of blood cells: red blood cells (RBCs), white blood cells (WBCs), and platelets. The CBC test yields numbers of these main types of cells, but also additional information, such as the volume of red blood cells, or hematocrit, and quantity of different types of white blood cells, the differential. Slide 3: CBCs are collected from patients in both inpatient and outpatient settings. Generally, 3 to 10 ml of whole blood are drawn from a peripheral vein directly into a tube. The tube must contain anticoagulant to prevent clots from obscuring the CBC analysis. Various types of anticoagulants are used including ethylenediaminetetraacetic acid (EDTA), heparin, and citrate. The most common anticoagulant used for the CBC is EDTA. -

Mature and Immature/Activated Cells Fractionation: Time for a Paradigm Shift in Differential Leucocyte Count Reporting?
diagnostics Article Mature and Immature/Activated Cells Fractionation: Time for a Paradigm Shift in Differential Leucocyte Count Reporting? Rana Zeeshan Haider 1,2,* , Najeed Ahmed Khan 3, Eloisa Urrechaga 4 and Tahir Sultan Shamsi 2 1 Baqai Institute of Hematology, Baqai Medical University, Karachi 75340, Pakistan 2 National Institute of Blood Disease (NIBD), Karachi 75300, Pakistan; [email protected] 3 Department of Computer Science, NED University of Engineering and Technology, Karachi 75270, Pakistan; [email protected] 4 Core Laboratory, Galdakao-Usansolo Hospital, 48960 Galdakao, Spain; [email protected] * Correspondence: [email protected]; Tel.: +92-343-507-1271 Abstract: Leucocytes, especially neutrophils featuring pro- and anti-cancerous characteristics, are involved in nearly every stage of tumorigenesis. Phenotypic and functional differences among mature and immature neutrophil fractions are well reported, and their correlation with tumor progression and therapy has emerging implications in modern oncology practices. Technological advancements enabled modern hematology analyzers to generate extended information (research parameters) during complete blood cell count (CBC) analysis. We hypothesized that neutrophil and lymphocyte fractions-related extended differential leucocytes count (DLC) parameters hold superior diagnostic utility over routine modalities. The present study was carried out over a four-and-a-half-year period wherein extended neutrophil (immature granulocyte [IG] and mature neutrophil [NEUT#&]), and lymphocyte (activated/high fluorescence lymphocyte count [HFLC] and resting lymphocyte Citation: Haider, R.Z.; Khan, N.A.; [LYMP#&]) parameters were challenged over routine neutrophil [NEUT#] and lymphocyte [LYMP#] Urrechaga, E.; Shamsi, T.S. Mature and Immature/Activated Cells items in a study population of 1067 hematological neoplasm patients. -

1 Blood Sampling and Blood Film Preparation and Examination
BCAC01 3/20/06 15:46 Page 1 1 Blood sampling and blood film preparation and examination Obtaining a blood specimen when he fainted at the end of a venepuncture and fell forward onto a hard floor, and two other Performing an accurate blood count and correctly patients, neither previously known to be epileptic, interpreting a blood film require that an appropriate who suffered epileptiform convulsions during vene- sample from the patient, mixed with the correct puncture. Such seizures may not be true epilepsy, amount of a suitable anticoagulant, is delivered to but consequent on hypoxia following brief vagal- the laboratory without undue delay. No artefacts induced cessation of heart beat [1]. If venepunctures should be introduced during these procedures. are being performed on children or on patients The identity of the patient requiring blood sam- unable to cooperate fully then the arm for vene- pling should be carefully checked before performing puncture should be gently but firmly immobilized a venepuncture. This is usually done by requesting by an assistant. Gloves should be worn during the patient to state surname, given name and date venepuncture, for the protection of the person of birth and, for hospital inpatients, by checking a carrying out the procedure. Non-latex gloves must wristband to verify these details and, in addition, the be available if either the phlebotomist or the patient hospital number. To reduce the chance of human is allergic to latex. The needle to enter the patient error, bottles should not be labelled in advance. The must not be touched, so that it remains sterile. -

Replacing the First Epidermal Growth Factor-Like Domain of Factor IX with That of Factor VII Enhances Activity in Vitro and in Canine Hemophilia B
Replacing the first epidermal growth factor-like domain of factor IX with that of factor VII enhances activity in vitro and in canine hemophilia B. J Y Chang, … , K M Brinkhous, H R Roberts J Clin Invest. 1997;100(4):886-892. https://doi.org/10.1172/JCI119604. Research Article Using the techniques of molecular biology, we made a chimeric Factor IX by replacing the first epidermal growth factor- like domain with that of Factor VII. The resulting recombinant chimeric molecule, Factor IXVIIEGF1, had at least a twofold increase in functional activity in the one-stage clotting assay when compared to recombinant wild-type Factor IX. The increased activity was not due to contamination with activated Factor IX, nor was it due to an increased rate of activation by Factor VIIa-tissue factor or by Factor XIa. Rather, the increased activity was due to a higher affinity of Factor IXVIIEGF1 for Factor VIIIa with a Kd for Factor VIIIa about one order of magnitude lower than that of recombinant wild- type Factor IXa. In addition, results from animal studies show that this chimeric Factor IX, when infused into a dog with hemophilia B, exhibits a greater than threefold increase in clotting activity, and has a biological half-life equivalent to recombinant wild-type Factor IX. Find the latest version: https://jci.me/119604/pdf Replacing the First Epidermal Growth Factor-like Domain of Factor IX with That of Factor VII Enhances Activity In Vitro and in Canine Hemophilia B Jen-Yea Chang,* Dougald M. Monroe,* Darrel W. Stafford,*‡ Kenneth M. -

Coagulation Simplified…
Coagulation Simplified… Published by ACKNOWLEDGEMENTS CONTENTS We gratefully acknowledge the support and funding provided by the Ontario Ministry of Health 1. The Basics of Coagulation and Clot Breakdown . 4–7 and Long-Term Care. 2. Routine Coagulation Tests . 8–17 Special thanks to the following people and organizations who provided their expertise in Evaluating coagulation in the laboratory . 8 reviewing the content of this handbook: Sample collection for coagulation testing . 9 Prothrombin Time (PT) . 10 L Gini Bourner (QMP-LS Hematology Committee) International Normalized Ratio (INR) . 11 L Dr. Jeannie Callum Activated Partial Thromboplastin Time (APTT) . 12 L Dr. Allison Collins Thrombin Time (TT) . 13 Fibrinogen . 14 L Dr. William Geerts D-dimer . 15 L Dr. Alejandro Lazo-Langner Anti-Xa assay . 16 L Dr. Ruth Padmore (QMP-LS Hematology Committee) Summary . 17 L Anne Raby (QMP-LS Hematology Committee) 3. Anticoagulant Drugs . 18–25 L Dr. Margaret Rand Unfractionated Hepari n (UFH) . 18 L Dr. Alan Tinmouth Low Molecular Weight Heparins (LMWHs) . 19 Fondaparinux . 20 Warfarin . 21 Thanks also to: Direct Thrombin Inhibitors (DTI) . 23 L Dale Roddick, photographer, Sunnybrook Health Sciences Centre Direct Xa Inhibitors . 25 L Reena Manohar, graphic artist, Sunnybrook Health Sciences Centre 4. Evaluating Abnormal Coagulation Tests . 26–29 L The ECAT Foundation Prolonged PT / INR with normal APTT . 26 CLOT-ED Images used or modified with permission from Prolonged APTT with normal PT / INR . 27 the ECAT Foundation, The Netherlands. Prolonged APTT and PT / INR . 28 Prolonged Thrombin Time (TT) with normal or prolonged APTT and PT / INR . 29 March 2013 5. Approach to the Evaluation of the Bleeding Patient .