How to Approach a Patient with Bleeding?
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WFH Treatment Guidelines 3Ed Chapter 7 Treatment Of
96 TREATMENT OF SPECIFIC 7 HEMORRHAGES Johnny Mahlangu1 | Gerard Dolan2 | Alison Dougall3 | Nicholas J. Goddard4 | Enrique D. Preza Hernández5 | Margaret V. Ragni6 | Bradley Rayner7 | Jerzy Windyga8 | Glenn F. Pierce9 | Alok Srivastava10 1 Department of Molecular Medicine and Haematology, University of the Witwatersrand, National Health Laboratory Service, Johannesburg, South Africa 2 Guy’s and St. Thomas’ Hospitals NHS Foundation Trust, London, UK 3 Special Care Dentistry Division of Child and Public Dental Health, School of Dental Science, Trinity College Dublin, Dublin Dental University Hospital, Dublin, Ireland 4 Department of Trauma and Orthopaedics, Royal Free Hospital, London, UK 5 Mexico City, Mexico 6 Division of Hematology/Oncology, Department of Medicine, University of Pittsburgh Medical Center, Pittsburgh, Pennsylvania, USA 7 Cape Town, South Africa 8 Department of Hemostasis Disorders and Internal Medicine, Laboratory of Hemostasis and Metabolic Diseases, Institute of Hematology and Transfusion Medicine, Warsaw, Poland 9 World Federation of Hemophilia, Montreal, QC, Canada 10 Department of Haematology, Christian Medical College, Vellore, India All statements identified as recommendations are • In general, the main treatment for bleeding episodes in consensus based, as denoted by CB. patients with severe hemophilia is prompt clotting factor replacement therapy and rehabilitation. However, different types of bleeds and bleeding at particular anatomical sites 7.1 Introduction may require more specific management with additional -

Crofab Brochure
Control With Confidence The only antivenom derived from native US pit vipers to treat envenomations from all species of North American pit vipers1 CroFab is the only antivenom Derived from geographically and clinically relevant US snakes for comprehensive coverage of all North American pit viper envenomations1 Designed with small, venom-specific protein (Fab) fragments for rapid neutralization of venom toxins throughout affected tissue1,2 With Level 1 evidence in the treatment of copperhead envenomation3 Manufactured to yield the highest level of quality, purity, and safety1 With a proven efficacy and safety profile, backed by >20 years of clinical experience1 Reliably supplied throughout the United States4 CroFab meets World Health Organization (WHO) guidelines for effective antivenom, utilizing venom from 4 clinically relevant pit viper species native to the United States.1,5 Indication CroFab® Crotalidae Polyvalent Immune Fab (Ovine) is a sheep-derived antivenin indicated for the management of adult and pediatric patients with North American crotalid envenomation. The term crotalid is used to describe the Crotalinae subfamily (formerly known as Crotalidae) of venomous snakes which includes rattlesnakes, copperheads and cottonmouths/water moccasins. Important Safety Information Contraindications Do not administer CroFab® to patients with a known history of hypersensitivity to any of its components, or to papaya or papain unless the benefits outweigh the risks and appropriate management for anaphylactic reactions is readily available. Warnings and Precautions Coagulopathy: In clinical trials, recurrent coagulopathy (the return of a coagulation abnormality after it has been successfully treated with antivenin), characterized by decreased fibrinogen, decreased platelets, and elevated prothrombin time, occurred in approximately half of the patients studied; one patient required re-hospitalization and additional antivenin administration. -

Familial Multiple Coagulation Factor Deficiencies
Journal of Clinical Medicine Article Familial Multiple Coagulation Factor Deficiencies (FMCFDs) in a Large Cohort of Patients—A Single-Center Experience in Genetic Diagnosis Barbara Preisler 1,†, Behnaz Pezeshkpoor 1,† , Atanas Banchev 2 , Ronald Fischer 3, Barbara Zieger 4, Ute Scholz 5, Heiko Rühl 1, Bettina Kemkes-Matthes 6, Ursula Schmitt 7, Antje Redlich 8 , Sule Unal 9 , Hans-Jürgen Laws 10, Martin Olivieri 11 , Johannes Oldenburg 1 and Anna Pavlova 1,* 1 Institute of Experimental Hematology and Transfusion Medicine, University Clinic Bonn, 53127 Bonn, Germany; [email protected] (B.P.); [email protected] (B.P.); [email protected] (H.R.); [email protected] (J.O.) 2 Department of Paediatric Haematology and Oncology, University Hospital “Tzaritza Giovanna—ISUL”, 1527 Sofia, Bulgaria; [email protected] 3 Hemophilia Care Center, SRH Kurpfalzkrankenhaus Heidelberg, 69123 Heidelberg, Germany; ronald.fi[email protected] 4 Department of Pediatrics and Adolescent Medicine, University Medical Center–University of Freiburg, 79106 Freiburg, Germany; [email protected] 5 Center of Hemostasis, MVZ Labor Leipzig, 04289 Leipzig, Germany; [email protected] 6 Hemostasis Center, Justus Liebig University Giessen, 35392 Giessen, Germany; [email protected] 7 Center of Hemostasis Berlin, 10789 Berlin-Schöneberg, Germany; [email protected] 8 Pediatric Oncology Department, Otto von Guericke University Children’s Hospital Magdeburg, 39120 Magdeburg, Germany; [email protected] 9 Division of Pediatric Hematology Ankara, Hacettepe University, 06100 Ankara, Turkey; Citation: Preisler, B.; Pezeshkpoor, [email protected] B.; Banchev, A.; Fischer, R.; Zieger, B.; 10 Department of Pediatric Oncology, Hematology and Clinical Immunology, University of Duesseldorf, Scholz, U.; Rühl, H.; Kemkes-Matthes, 40225 Duesseldorf, Germany; [email protected] B.; Schmitt, U.; Redlich, A.; et al. -

Immune-Pathophysiology and -Therapy of Childhood Purpura
Egypt J Pediatr Allergy Immunol 2009;7(1):3-13. Review article Immune-pathophysiology and -therapy of childhood purpura Safinaz A Elhabashy Professor of Pediatrics, Ain Shams University, Cairo Childhood purpura - Overview vasculitic disorders present with palpable Purpura (from the Latin, purpura, meaning purpura2. Purpura may be secondary to "purple") is the appearance of red or purple thrombocytopenia, platelet dysfunction, discolorations on the skin that do not blanch on coagulation factor deficiency or vascular defect as applying pressure. They are caused by bleeding shown in table 1. underneath the skin. Purpura measure 0.3-1cm, A thorough history (Table 2) and a careful while petechiae measure less than 3mm and physical examination (Table 3) are critical first ecchymoses greater than 1cm1. The integrity of steps in the evaluation of children with purpura3. the vascular system depends on three interacting When the history and physical examination elements: platelets, plasma coagulation factors suggest the presence of a bleeding disorder, and blood vessels. All three elements are required laboratory screening studies may include a for proper hemostasis, but the pattern of bleeding complete blood count, peripheral blood smear, depends to some extent on the specific defect. In prothrombin time (PT) and activated partial general, platelet disorders manifest petechiae, thromboplastin time (aPTT). With few exceptions, mucosal bleeding (wet purpura) or, rarely, central these studies should identify most hemostatic nervous system bleeding; -

We Get Biased on Things That Make Sense
DIALOGUE AND DISCUSSION We Get Biased on Things That Make Sense MERIH T. TESFAZGHI When I was teaching third year medical students, and bench- reaction might be characterized by a marked left shift lecturing clinical laboratory science students, I emphasized which may display a range of immature cells that are also the importance of clinical details and the history of patients seen in CML. Severe left shift especially in the absence of in laboratory test processing, especially in the evaluation of evident infection (or other underlying diseases) may peripheral blood films. During those times, one of my strongly suggest direct bone marrow involvement. In students stated, “but including details of patients might such scenario, clinical details of patients such as absence somehow affect the decision of the reviewer, and may lead to or presence of enlargement of extramedullar organs is Downloaded from a bias.” I responded that we are biased on things that make highly sought. sense. This means that we decide based on the evidence we see from a microscopy evaluation in the light of the clinical Ø The coexistence of multiple conditions in a given detail and history. How do clinical details and history help us patient. One condition may mask the presence of the reach decisions? other condition. For example, the coexistence of megaloblastic anemia with iron deficiency anemia. http://hwmaint.clsjournal.ascls.org/ If specimens are the “in vitro ambassadors” of patients to the Usually, megaloblastic anemia is characterized by laboratory, then properly completed request forms are their increased size of red blood cells (MCV), but when “credentials”. -

Haemostatic Problems in Liver Disease
Gut: first published as 10.1136/gut.27.3.339 on 1 March 1986. Downloaded from Gut, 1986, 27, 339-349 Progress report Haemostatic problems in liver disease The liver plays a major role in the control of coagulation and as a result haemostatic problems are detected in approximately 75% of patients with liver disease.1 The coagulation abnormalities are both complex and multifactorial and depend on the balance between hepatic synthesis and clearance of activated coagulation proteins and their inhibitors; the presence or absence of dysfibrinogenaemia; thrombocytopenia, abnormal platelet function, and disseminated intravascular coagulation. Some patients will present with petechiae, ecchymosis or epistaxis, but most patients are asymptomatic or only bleed after venepuncture or liver biopsy. Alternatively haemorrhage may be life threatening and patients may die from variceal bleeding or from disseminated intravascular coagulation. The reasons for this disparity are not yet clear, but after the introduction of newer techniques, in particular the development of immunological assays for the antigens of coagulation proteins, our understanding of these problems has improved. The normal coagulation and fibrinolytic systems are depicted in Figures 1 and 2 while the major .__Intrinsic___ _ pathwY http://gut.bmj.com/ Kallikrein.o- PK | HMWKq 8t XII -*xiiXIIa_4------- ATIII ~ ~ 'I L1HMWK - XI* Xla %' xC-a; --------- -- on September 28, 2021 by guest. Protected copyright. IX - IXa VII -e'VIIca Extrinsic pathway [X VIII a Ce X P'okin C ATIII Ca+ XIII Common mI ~V PL II a pathway I XIIIa Fibrinogen - Fibrin Fig. 1 The coagulation cascade. HMWK=high molecular weight Kinogen, PK=Pre-Kallikrein, A TIII=antithrornbin III, PL=platelets, Ca" = Calcium, TF=tissue factor, -t- =proteolytic activation, -+=conversion ofcoagulation protein, -- -+=inhibition by plasma inhibitors, tit =crosslinking, a=activated coagulation enzyme. -

The Underrecognized Prothrombotic Vascular Disease of COVID-19
Journal Articles 2020 The underrecognized prothrombotic vascular disease of COVID-19. KP Cohoon G Mahé AC Spyropoulos Zucker School of Medicine at Hofstra/Northwell, [email protected] Follow this and additional works at: https://academicworks.medicine.hofstra.edu/articles Part of the Internal Medicine Commons Recommended Citation Cohoon K, Mahé G, Spyropoulos A. The underrecognized prothrombotic vascular disease of COVID-19.. 2020 Jan 01; 4(5):Article 6487 [ p.]. Available from: https://academicworks.medicine.hofstra.edu/articles/ 6487. Free full text article. This Article is brought to you for free and open access by Donald and Barbara Zucker School of Medicine Academic Works. It has been accepted for inclusion in Journal Articles by an authorized administrator of Donald and Barbara Zucker School of Medicine Academic Works. For more information, please contact [email protected]. Received: 6 May 2020 | Revised: 16 May 2020 | Accepted: 21 May 2020 DOI: 10.1002/rth2.12396 LETTER TO THE EDITOR The underrecognized prothrombotic vascular disease of COVID-19 We have read with interest “COVID-19-associated coagulopathy around elevated markers of hypercoagulability, including D-dimer, and thromboembolic disease: Commentary on an interim expert tissue factor expression, fibrinogen levels, factor VIII levels, guidance” recently provided by Cannegieter and Klok.1 This com- short-activated partial thromboplastin time, platelet binding, and mentary exemplifies the importance that venous thromboembolism thrombin formation.8 Based on well-defined clinical and laboratory (VTE) and atheroembolism may be underrepresented and a cause parameters, a proposal for staging COVID-19 coagulopathy may for increased morbidity and mortality among coronavirus disease provide treatment algorithms stratified into 3 stages.9 However, 2019 (COVID-19) patients. -
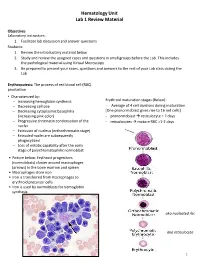
Hematology Unit Lab 1 Review Material
Hematology Unit Lab 1 Review Material Objectives Laboratory instructors: 1. Facilitate lab discussion and answer questions Students: 1. Review the introductory material below 2. Study and review the assigned cases and questions in small groups before the Lab. This includes the pathological material using Virtual Microscopy 3. Be prepared to present your cases, questions and answers to the rest of your Lab class during the Lab Erythropoiesis: The process of red blood cell (RBC) production • Characterized by: − Increasing hemoglobin synthesis Erythroid maturation stages (Below): − Decreasing cell size - Average of 4 cell divisions during maturation − Decreasing cytoplasmic basophilia [One pronormoblast gives rise to 16 red cells] (increasing pink color) - pronormoblast → reticulocyte = 7 days − Progressive chromatin condensation of the - reticulocytes → mature RBC =1-2 days nuclei − Extrusion of nucleus (orthochromatic stage) − Extruded nuclei are subsequently phagocytized − Loss of mitotic capability after the early stage of polychromatophilic normoblast • Picture below: Erythroid progenitors (normoblasts) cluster around macrophages (arrows) in the bone marrow and spleen • Macrophages store iron • Iron is transferred from macrophages to erythroid precursor cells • Iron is used by normoblasts for hemoglobin synthesis aka nucleated rbc aka reticulocyte 1 Mature Red Blood Cell 7-8 microns; round / ovoid biconcave disc with orange-red cytoplasm, no RNA, no nucleus; survives ~120 days in circulation Classification of Anemia by Morphology 1. -

Copyrighted Material
CHAPTER 1 The Blood Film and Count Blood Blood is a life‐sustaining fluid that circulates through the heart and blood vessels. It carries oxygen and nutrients to the tissues and waste products to the lungs, liver and kidneys, where they can be removed from the body. Usually when blood is removed from the body it forms a solid blood clot. However, if clotting is prevented by mixing with an anticoagulant, the blood separates, under the influence of gravity, into three layers (Fig. 1.1). The bottom layer is deep red in colour and is composed of red cells. The top layer is clear and pale yellow. It is called plasma and is composed of various salts and proteins dissolved in water. In between is a narrow layer called the buffy coat because of its buff or yellowish white colour. The buffy coat is composed mainly of cells of a variety of types, collectively known as white cells. In addition there are small cellular fragments, called platelets, which have a role in blood clotting. The bloodCOPYRIGHTED film MATERIAL Although we can judge the proportions of red cells and white cells in a tube of sedimented blood, we get far more information if the blood is carefully mixed and a thin layer is spread on a glass A Beginner’s Guide to Blood Cells, Third Edition. Barbara J. Bain. © 2017 John Wiley & Sons Ltd. Published 2017 by John Wiley & Sons Ltd. 1 0003049575.indd 1 2/24/2017 9:50:31 AM 2 A Beginner’s Guide to Blood Cells Plasma Buffy coat Red cells Fig. -

Blood Film Preparation and Staining Procedures
INTERPRETATION OF THE PERIPHERAL 00. ם BLOOD FILM 0272–2712/02 $15.00 BLOOD FILM PREPARATION AND STAINING PROCEDURES Berend Houwen, MD, PhD The blood film is one of the world’s most widely and frequently used tests and has undergone remarkably few changes since its introduction as a clinical diagnostic tool in the late 1800s. The origins of stained blood film microscopy are somewhat obscure, and a clear reference for the ‘‘first person or persons’’ to describe its use as a clinical laboratory procedure cannot be established with certainty. Antonie van Leeuwenhoek, in the seventeenth century, was the first to describe blood cells, using whole blood preparations and not blood films for his observations on blood corpuscles. In fact, he would not have been able to use blood film–based microscopy because it requires considerably more sophisti- cated optics than were available in his day. A second technologic innovation enabling blood film microscopy was the introduction of aniline dyes in the second half of the nineteenth century. This made it possible to study individual blood cells by light microscopy after a small amount of blood had been placed and smeared onto glass slides, dried, fixed, and then stained. Paul Ehrlich introduced eosin as the first of these dyes for staining blood films in 1856, followed by hematoxylin in 1865, and later by the metachromatic Romanowsky dyes. A few refinements have since been made, consisting mainly of improve- ments in dye quality, staining procedures, automation of slide preparation, and staining, but the basic elements of blood film preparation and analysis have not changed for over a century. -

Guidelines for the Management of Haemophilia in Australia
Guidelines for the management of haemophilia in Australia A joint project between Australian Haemophilia Centre Directors’ Organisation, and the National Blood Authority, Australia © Australian Haemophilia Centre Directors’ Organisation, 2016. With the exception of any logos and registered trademarks, and where otherwise noted, all material presented in this document is provided under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Australia (http://creativecommons.org/licenses/by-nc-sa/3.0/au/) licence. You are free to copy, communicate and adapt the work for non-commercial purposes, as long as you attribute the authors and distribute any derivative work (i.e. new work based on this work) only under this licence. If you adapt this work in any way or include it in a collection, and publish, distribute or otherwise disseminate that adaptation or collection to the public, it should be attributed in the following way: This work is based on/includes the Australian Haemophilia Centre Directors’ Organisation’s Guidelines for the management of haemophilia in Australia, which is licensed under the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Australia licence. Where this work is not modified or changed, it should be attributed in the following way: © Australian Haemophilia Centre Directors’ Organisation, 2016. ISBN: 978-09944061-6-3 (print) ISBN: 978-0-9944061-7-0 (electronic) For more information and to request permission to reproduce material: Australian Haemophilia Centre Directors’ Organisation 7 Dene Avenue Malvern East VIC 3145 Telephone: +61 3 9885 1777 Website: www.ahcdo.org.au Disclaimer This document is a general guide to appropriate practice, to be followed subject to the circumstances, clinician’s judgement and patient’s preferences in each individual case. -

Complete Blood Count in Primary Care
Complete Blood Count in Primary Care bpac nz better medicine Editorial Team bpacnz Tony Fraser 10 George Street Professor Murray Tilyard PO Box 6032, Dunedin Clinical Advisory Group phone 03 477 5418 Dr Dave Colquhoun Michele Cray free fax 0800 bpac nz Dr Rosemary Ikram www.bpac.org.nz Dr Peter Jensen Dr Cam Kyle Dr Chris Leathart Dr Lynn McBain Associate Professor Jim Reid Dr David Reith Professor Murray Tilyard Programme Development Team Noni Allison Rachael Clarke Rebecca Didham Terry Ehau Peter Ellison Dr Malcolm Kendall-Smith Dr Anne Marie Tangney Dr Trevor Walker Dr Sharyn Willis Dave Woods Report Development Team Justine Broadley Todd Gillies Lana Johnson Web Gordon Smith Design Michael Crawford Management and Administration Kaye Baldwin Tony Fraser Kyla Letman Professor Murray Tilyard Distribution Zane Lindon Lyn Thomlinson Colleen Witchall All information is intended for use by competent health care professionals and should be utilised in conjunction with © May 2008 pertinent clinical data. Contents Key points/purpose 2 Introduction 2 Background ▪ Haematopoiesis - Cell development 3 ▪ Limitations of reference ranges for the CBC 4 ▪ Borderline abnormal results must be interpreted in clinical context 4 ▪ History and clinical examination 4 White Cells ▪ Neutrophils 5 ▪ Lymphocytes 9 ▪ Monocytes 11 ▪ Basophils 12 ▪ Eosinophils 12 ▪ Platelets 13 Haemoglobin and red cell indices ▪ Low haemoglobin 15 ▪ Microcytic anaemia 15 ▪ Normocytic anaemia 16 ▪ Macrocytic anaemia 17 ▪ High haemoglobin 17 ▪ Other red cell indices 18 Summary Table 19 Glossary 20 This resource is a consensus document, developed with haematology and general practice input. We would like to thank: Dr Liam Fernyhough, Haematologist, Canterbury Health Laboratories Dr Chris Leathart, GP, Christchurch Dr Edward Theakston, Haematologist, Diagnostic Medlab Ltd We would like to acknowledge their advice, expertise and valuable feedback on this document.