Tio2 Photocatalysis in Portland Cement Systems: Fundamentals of Self Cleaning Effect and Air Pollution Mitigation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

White Concrete & Colored Concrete Construction
WHITE CEMENT CONCRETE AND COLORED CONCRETE CONSTRUCTION and developers Owners seeking dynamic architectural results demand the benefits that only white or colored concrete can deliver. Informed designers rely on careful selection of materials and attention to mixing and placement details to achieve eye-catching architectural and decorative finishes. Because of its versatility, concrete offers endless design possibilities. A wide variety of decorative looks are achieved by using colored aggregates and pigments, and by varying surface finishes, treatments, and textures. Combining white cement with pigments and colored aggregates expands the number of colors available to discerning customers. Colors are cleaner and more intense because pigments and specialty Figure 1. The Condell Medical Center is built with load-bearing precast concrete aggregates don’t have to overcome the panels with an architectural finish achieved by using white cement. grayish paste of common concrete. Courtesy of Condell Medical Center. Mix Designs • CSA A3000 cements that conform to the Canadian From a structural design perspective, white cement-based Standards Association standards for Cementitious Materials concrete mixes are identical to gray cement-based mixes. Mix • American Association of State Highway and Transportation designs for white or colored concrete differ from common Officials, AASHTO M85 Standard Specification for Portland concrete in that they are formulated based on each ingredient’s Cement effect on concrete color. Listed in order of decreasing -
Cement Data Sheet
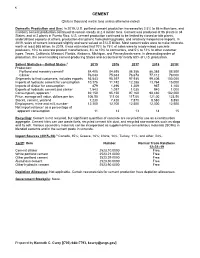
42 CEMENT (Data in thousand metric tons unless otherwise noted) Domestic Production and Use: In 2019, U.S. portland cement production increased by 2.5% to 86 million tons, and masonry cement production continued to remain steady at 2.4 million tons. Cement was produced at 96 plants in 34 States, and at 2 plants in Puerto Rico. U.S. cement production continued to be limited by closed or idle plants, underutilized capacity at others, production disruptions from plant upgrades, and relatively inexpensive imports. In 2019, sales of cement increased slightly and were valued at $12.5 billion. Most cement sales were to make concrete, worth at least $65 billion. In 2019, it was estimated that 70% to 75% of sales were to ready-mixed concrete producers, 10% to concrete product manufactures, 8% to 10% to contractors, and 5% to 12% to other customer types. Texas, California, Missouri, Florida, Alabama, Michigan, and Pennsylvania were, in descending order of production, the seven leading cement-producing States and accounted for nearly 60% of U.S. production. Salient Statistics—United States:1 2015 2016 2017 2018 2019e Production: Portland and masonry cement2 84,405 84,695 86,356 86,368 88,500 Clinker 76,043 75,633 76,678 77,112 78,000 Shipments to final customers, includes exports 93,543 95,397 97,935 99,406 100,000 Imports of hydraulic cement for consumption 10,376 11,742 12,288 13,764 15,000 Imports of clinker for consumption 879 1,496 1,209 967 1,100 Exports of hydraulic cement and clinker 1,543 1,097 1,035 940 1,000 Consumption, apparent3 92,150 95,150 97,160 98,480 102,000 Price, average mill value, dollars per ton 106.50 111.00 117.00 121.00 123.50 Stocks, cement, yearend 7,230 7,420 7,870 8,580 8,850 Employment, mine and mill, numbere 12,300 12,700 12,500 12,300 12,500 Net import reliance4 as a percentage of apparent consumption 11 13 13 14 15 Recycling: Cement is not recycled, but significant quantities of concrete are recycled for use as a construction aggregate. -

Best Practice Guidelines for Selecting Concrete Bridge Deck Sealers
BEST PRACTICE GUIDELINES FOR SELECTING CONCRETE BRIDGE DECK SEALERS GENERAL Penetrating sealers are used on highway Portland Cement Concrete bridge decks to reduce the rate of chloride attack on the reinforcing steel corrosion thereby extending service life and reducing life-cycle structure costs of the bridge deck (preventative maintenance). By applying penetrating sealers to existing concrete surfaces, the permeability of the concrete is reduced by up to one order of magnitude. The permeability of the concrete is one of the most important factors which will effect the rate of deterioration of rebar corrosion, alkali-aggregate reaction, carbonation, and the effects of freeze-thaw cycles of which all could occur at the same time. HISTORY Concrete sealing has a long history in Alberta; in the 1960’s boiled linseed oil was used for maintenance of existing bridge curbs. Epoxies and other types of sealers were also tried in the 60’s. The first epoxy-wearing surface in Alberta was in 1963 at Morley. Dehydratine, a black tarry substance, was routinely used on abutments, since the late 1960’s. Epoxy and acrylic sealers were routinely used on standard precast girders starting in the middle of 1970’s. Penetrating Silane sealers were first used in Alberta on concrete bridge decks in 1986. CONCRETE PROPERTIES • Concrete is a conglomerate of sand and rock glued together by cement paste (hydrated cement – cement reacted with water to form calcium silicate chemical bonds). • Concrete is a synergistic material; the whole has greater structural properties -

General Product Catalogue
Solutions In Concrete Construction GENERAL PRODUCT CATALOGUE Other Available Catalogues • Concrete Forming Systems • Bridge Deck Forming and Hanging Systems • Precast Concrete Products • Concrete Anchoring Systems • Rock Anchoring and Bolt Systems www.nca.ca Call Us Toll Free: 1-888-777-9272 LOCATIONS Alberta Western Division Calgary Edmonton Red Deer 14760 - 116th Avenue NW 3834 54th Avenue S. E. 14760 - 116th Avenue NW #5, 7803 - 50 Avenue With over 45 years of experience, National Concrete Accessories (NCA), is Canada’s Edmonton, AB T5M3G1 Calgary, AB T2C2K9 Edmonton, AB T5M3G1 Red Deer, AB T4P1M8 leading manufacturer and distributor of concrete accessories. With 14 branches across Tel: (780) 451-1212 Tel: (403) 279-7089 Tel: (780) 451-1212 Tel: (403) 342-0210 Canada, including manufacturing plants in Toronto and Edmonton, we are your one-stop Fax: (780) 455-0827 Fax: (403) 279-4397 Fax: (780) 455-0827 Fax: (403) 341-3245 shop for concrete form hardware, accessories and construction products. Specializing in product variety, quality and superior service, we distribute a complete line of construction British Columbia and restoration products, including tools & equipment, decorative concrete, and building envelope products. NCA works in partnership with premium brands such as, Acrow- Burnaby Kamloops Kelowna Prince George Richmond, DOW, Sika, Makita, W.R. Meadows, Bosch, Mapei, Titebond, UCAN, 7885 Venture Street 855 Laval Crescent 1948 Dayton Street #101- 2050 Robertson Road Husqvarna, Xypex, Butterfield Color, Marshalltown and Chapin to provide you with the Burnaby, BC V5A1V1 Kamloops, BC V2C5P2 Kelowna, BC V1Y7W6 Prince George, BC V2N1X6 Tel: (604) 435-6700 Tel: (250) 374-6295 Tel: (250) 717-1616 Tel: (250) 614-1212 best cost-effective solutions to meet your concrete job requirements. -

CEMENT for BUILDING with AMBITION Aalborg Portland A/S Portland Aalborg Cover Photo: the Great Belt Bridge, Denmark
CEMENT FOR BUILDING WITH AMBITION Aalborg Portland A/S Cover photo: The Great Belt Bridge, Denmark. AALBORG Aalborg Portland Holding is owned by the Cementir Group, an inter- national supplier of cement and concrete. The Cementir Group’s PORTLAND head office is placed in Rome and the Group is listed on the Italian ONE OF THE LARGEST Stock Exchange in Milan. CEMENT PRODUCERS IN Cementir's global organization is THE NORDIC REGION divided into geographical regions, and Aalborg Portland A/S is included in the Nordic & Baltic region covering Aalborg Portland A/S has been a central pillar of the Northern Europe. business community in Denmark – and particularly North Jutland – for more than 125 years, with www.cementirholding.it major importance for employment, exports and development of industrial knowhow. Aalborg Portland is one of the largest producers of grey cement in the Nordic region and the world’s leading manufacturer of white cement. The company is at the forefront of energy-efficient production of high-quality cement at the plant in Aalborg. In addition to the factory in Aalborg, Aalborg Portland includes five sales subsidiaries in Iceland, Poland, France, Belgium and Russia. Aalborg Portland is part of Aalborg Portland Holding, which is the parent company of a number of cement and concrete companies in i.a. the Nordic countries, Belgium, USA, Turkey, Egypt, Malaysia and China. Additionally, the Group has acti vities within extraction and sales of aggregates (granite and gravel) and recycling of waste products. Read more on www.aalborgportlandholding.com, www.aalborgportland.dk and www.aalborgwhite.com. Data in this brochure is based on figures from 2017, unless otherwise stated. -

Gray Portland Cement and Cement Clinker from Japan
Gray Portland Cement and Cement Clinker from Japan Investigation No. 731-TA-461 (Fourth Review) Publication 4704 June 2017 U.S. International Trade Commission Washington, DC 20436 U.S. International Trade Commission COMMISSIONERS Rhonda K. Schmidtlein, Chairman David S. Johanson, Vice Chairman Irving A. Williamson Meredith M. Broadbent F. Scott Kieff Catherine DeFilippo Director of Operations Staff assigned Carolyn Carlson, Investigator Gregory LaRocca, Industry Analyst Andrew Knipe, Economist Heng Loke, Attorney Fred Ruggles, Supervisory Investigator Address all communications to Secretary to the Commission United States International Trade Commission Washington, DC 20436 U.S. International Trade Commission Washington, DC 20436 www.usitc.gov Gray Portland Cement and Cement Clinker from Japan Investigation No. 731-TA-461 (Fourth Review) Publication 4704 June 2017 CONTENTS Page Determination .............................................................................................................................1 Views of the Commission .............................................................................................................3 Information obtained in the review ........................................................................................... I-1 Background ...................................................................................................................................... I-1 Responses to the Commission's notice of institution ..................................................................... -

Ts-63-34.00-001
IDM UID YHM3FJ VERSION CREATED ON / VERSION / STATUS 09 Jun 2020 / 2.1 / Approved EXTERNAL REFERENCE / VERSION Technical Specifications (In-Cash Procurement) TS-63-34.00-001 - IOTS - 000001 : Technical Specifications B50s Grouting and related works This document provides requirements for grouting works and accompanied surface preparation and oil-resistant painting works to be performed on IO platform in B50s (B51/B52/A53) in order to complete the interface between PBS63 and each of PBS34.10 and PBS34.40. PDF generated on 15 Jun 2020 DISCLAIMER : UNCONTROLLED WHEN PRINTED – PLEASE CHECK THE STATUS OF THE DOCUMENT IN IDM ITER_D_YHM3FJ v2.1 Table of Contents 1 PURPOSE ............................................................................................................................ 2 2 SCOPE ................................................................................................................................. 2 3 DEFINITIONS .................................................................................................................... 2 4 DURATION ......................................................................................................................... 3 5 WORK DESCRIPTION ..................................................................................................... 3 5.1 Scope of Works ............................................................................................................. 3 5.2 Grouting volume and preparation work ................................................................... -

GENERAL 1.01 DESCRIPTION the WORK of This Section Includes Providing Concrete Formwork, Bracing
SECTION 03100 FORMWORK PART 1 -- GENERAL 1.01 DESCRIPTION The WORK of this Section includes providing concrete formwork, bracing, shoring, supports and formwork requirements for concrete construction. 1.02 RELATED SECTIONS The WORK of the following Sections applies to the WORK of this Section. Other Sections of the Specifications, not referenced below, shall also apply to the extent required for proper performance of the WORK. 1. Section 03200 Concrete Reinforcement 2. Section 03290 Joints in Concrete Structure 3. Section 03300 Cast-in-Place Concrete 4. Section 03600 Grout 1.03 STANDARD SPECIFICATIONS Except as otherwise indicated in this Section of the Specifications, the CONTRACTOR shall comply with the Standard Specifications for Public Works Construction (SSPWC), as specified in Section 01071 Standard References. 1.04 QUALITY ASSURANCE A. REFERENCES: This section contains references to the documents listed below. They are a part of this section as specified and modified. Where a referenced document cites other standards, such standards are included as references under this section as if referenced directly. In the event of conflict between the requirements of this section and those of the listed documents, the requirements of this section shall prevail. Unless otherwise specified, references to documents shall mean the documents in effect at the time of Advertisement for Bids or Invitation to Bid (or on the effective date of the Agreement if there were no Bids). If referenced documents have been discontinued by the issuing organization, references to those documents shall mean the replacement documents FORMWORK DECEMBER 2014 PWS CONTRACT NO. 13-40UTIL 03100-1 issued or otherwise identified by that organization or, if there are no replacement documents, the last version of the document before it was discontinued. -

Recovery of Waste Expanded Polystyrene in Lightweight Concrete Production
73 The Mining-Geology-Petroleum Engineering Bulletin Recovery of waste expanded polystyrene UDC: 624.01 in lightweight concrete production DOI: 10.17794/rgn.2019.3.8 Original scientifi c paper Gordan Bedeković1; Ivana Grčić2; Aleksandra Anić Vučinić2; Vitomir Premur2 1University of Zagreb, Faculty of Mining, Geology and Petroleum Engineering, Pierottijeva 6, 10000 Zagreb, Croatia 2University of Zagreb, Faculty of Geotechnical Engineering, Hallerova 7, 42000 Varaždin, Croatia Abstract Polystyrene concrete, as a type of lightweight aggregate concrete, has been used in civil construction for years. The use of waste expanded polystyrene (EPS) as a fi ll material in lightweight concrete production is highly recommended from the point of view of the circular economy. Published data shows that an increase in the proportion of lightweight aggre- gates, i.e. EPS, results in a decrease in strength, bulk density and thermal conductivity of the concrete. Utilizing large quantities of waste EPS in non-structural polystyrene concrete production is particularly important. Unlike structural polystyrene concrete, according to the published papers, non-structural polystyrene concrete has not been investigated suffi ciently. The purpose of this paper is to determine the infl uence of the ratios of the basic components in a concrete mixture on the bulk density and compressive strength of non-structural polystyrene concrete produced by utilizing waste EPS as a fi ll material. The test specimens, i.e. cubes with an edge length of 100 mm each, were prepared in labora- tory conditions by varying the proportions of EPS, sand up to 600 g and cement ranging from 300 g to 450 g per speci- men. -

Portland Cements CAS Reg
3251 Bath Pike Nazareth, PA 18064 MATERIAL SAFETY DATA SHEET Section 1 - IDENTIFICATION Product Name: Portland Cements CAS Reg. No.: 65997-15-1 Chemical Name and Synonyms: Portland Cement, Cement, Hydraulic Cement Trade Names: Portland Cement – Types I, IA, II, III, IIIA, GU, MS,LH, HE; SAYLOR’S Portland Types: I, IA, II, III; PRONTO, Flamingo Brixment White Portland Cement; Oil Well Cement Class A, H. MSDS Information: This MSDS supersedes prior MSDS’s for the products noted above. This MSDS covers a number of products with similar applications and occupational exposure hazards. Specific constituents and methods of preparation for these products will vary. The term “Portland Cement”, used in the text of this MSDS, refers to the above named products collectively. Chemical Family: Calcium silicate compounds; calcium compounds containing iron and aluminum; and gypsum are the primary constituents of these products. Informational Phone Numbers: (800) 437-7762 Customer Service - Nazareth, PA (800) 336-0366 Customer Service - Speed, IN (800) 624-8986 Customer Service - Martinsburg, WV (800) 386-2111 Customer Service - Mississauga, ONT Emergency Contact Information: (800)-424-9300 Chemtrec MSDS Prepared by: Essroc MSDS Development Committee - (610) 837-6725 – May 9, 2013 Section 2 - COMPONENTS Hazardous Ingredients: Component CAS No. OSHA PEL (8-hour TWA) ACGIH TLV Other Information Portland Cement 65997-15-1 15 mg total dust/m3 1.0 mg/m3 IDLH: 5000 mg/m3 3 5 mg respirable dust/m respirable LD50: No Data Gypsum 13397-24-5 15 mg total dust/m3 -

Armorhab: Design Reference Architecture (DRA) for Human
Copyright © 2016 by Dark Sea Industries LLC and the University of New Mexico. Published by The Mars Society with permission ArmorHab: Design Reference Architecture (DRA) for human habitation in deep space Peter Vorobieff Professor and Assistant Chair, Assistant Chair of Facilities, Department of Mechanical Engineering University of New Mexico, Albuquerque, NM, 87131 Phone: (505) 277-8347 email: [email protected] Affiliation: University of New Mexico Craig Davidson Administrator 4808 Downey NE Albuquerque, NM 87109 Phone: 505-720-2321 email: [email protected] Affiliation: Dark Sea Industries LLC Dr. Mahmoud Reda Taha, Peng Professor and Chair, Department of Civil Engineering University of New Mexico, Albuquerque, NM 87131-0001 Office: (505) 277-1258, Cell: (505) 385-8930, Fax (505) 277-1988 http://civil.unm.edu/faculty-staff/faculty-profiles/mahmoud-taha.html www.unm.edu/~mrtaha/index.htm Affiliation: University of New Mexico Christos Christodoulou Associate Dean for Research Distinguished Professor, Electrical & Computer Engineering University of New Mexico Albuquerque, NM 87131 Tel: (505) 277-6580 www.ece.unm.edu/faculty/cgc www.cosmiac.org Affiliation: University of New Mexico Anil K. Prinja Professor and Chair Department of Nuclear Engineering University of New Mexico Albuquerque, NM 87131-1070 -1- Copyright © 2016 by Dark Sea Industries LLC and the University of New Mexico. Published by The Mars Society with permission Phone: (505)-277-4600, Fax: (505)-277-5433 [email protected] Affiliation: University of New Mexico Svetlana V. Poroseva Assistant Professor Department of Mechanical Engineering University of New Mexico, Albuquerque, NM, 87131 Phone: 1(505) 277-1493, Fax: 1(505) 277-1571 email: poroseva at unm.edu Affiliation: University of New Mexico Mehran Tehrani Assistant Professor Department of Mechanical Engineering University of New Mexico, Albuquerque, NM, 87131 Phone: 1(505) 277-1493, Fax: 1(505) 277-1571 email: [email protected] Affiliation: University of New Mexico David T. -

HUMBOLDT-CATALOG-2017 COVER.Indd
Air Meters 140-143 Beam Testing 151 Compression Testing 162-171 Consistency 147 Corrosion 178-180 Crack Monitors 190-191 Cube Molds 152 Cylinder Capping 160-161 Cylinder Molds 153 Cylinder Testing 154-155 Cylinder Prep 158-159 Freeze-Thaw 186-187 Ground Penetrating Radar 176 Humidifiers 156 Humidity 157 Initial Set 146 Linear Traverse 192 Maturity 148 Mixers 150 Moisture (Slabs) 188-189 Pull-Off 175 Pulse Echo 183 Rebar Locators 176-177 Rebound Test Hammers 172-173 Resistivity 184-185 Self Consolidating Concrete 149 Slump 144-145 Strain 192 Strength 174 Ultrasonic 181-183 Unit Weight 143 CONCRETE 140 Air Meters H-2784.500 H-2783 H-2784 Humboldt Super Air Meter bubbles meets the recommendations of the ACI piece, self-locking clamps that quickly seal the lid ASTM C231, AASHTO T152 201 Concrete Durability Committee. to the base with proper tension aided by an o-ring The H-2784 Super Air Meter (SAM) is a testing This new air meter has been investigated using to assure a watertight seal. A large, easy-to-read, device that measures both the air void spacing and more than 300 lab and field mixtures at Oklahoma 4-inch diameter, direct percentage gauge with air volume of plastic (fresh) concrete in about 10 State University and the FHWA Turner Fairbanks calibration adjustments is accurate to the nearest minutes. Air void spacing has been shown to be Laboratories. As part of an ongoing Pooled Fund 0.1%. The bucket features cast handles, which a better indicator of concrete freeze-thaw dura- Study, the SAM is being used by 10 different DOTs improve usability, especially when using bucket as bility than total air content; however, until now, it on field concrete.