Iaspp Mediates P53 Selectivity Through a Modular Mechanism Fine-Tuning DNA Recognition
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells
Leukemia (2010) 24, 756–764 & 2010 Macmillan Publishers Limited All rights reserved 0887-6924/10 $32.00 www.nature.com/leu ORIGINAL ARTICLE Deregulated gene expression pathways in myelodysplastic syndrome hematopoietic stem cells A Pellagatti1, M Cazzola2, A Giagounidis3, J Perry1, L Malcovati2, MG Della Porta2,MJa¨dersten4, S Killick5, A Verma6, CJ Norbury7, E Hellstro¨m-Lindberg4, JS Wainscoat1 and J Boultwood1 1LRF Molecular Haematology Unit, NDCLS, John Radcliffe Hospital, Oxford, UK; 2Department of Hematology Oncology, University of Pavia Medical School, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy; 3Medizinische Klinik II, St Johannes Hospital, Duisburg, Germany; 4Division of Hematology, Department of Medicine, Karolinska Institutet, Stockholm, Sweden; 5Department of Haematology, Royal Bournemouth Hospital, Bournemouth, UK; 6Albert Einstein College of Medicine, Bronx, NY, USA and 7Sir William Dunn School of Pathology, University of Oxford, Oxford, UK To gain insight into the molecular pathogenesis of the the World Health Organization.6,7 Patients with refractory myelodysplastic syndromes (MDS), we performed global gene anemia (RA) with or without ringed sideroblasts, according to expression profiling and pathway analysis on the hemato- poietic stem cells (HSC) of 183 MDS patients as compared with the the French–American–British classification, were subdivided HSC of 17 healthy controls. The most significantly deregulated based on the presence or absence of multilineage dysplasia. In pathways in MDS include interferon signaling, thrombopoietin addition, patients with RA with excess blasts (RAEB) were signaling and the Wnt pathways. Among the most signifi- subdivided into two categories, RAEB1 and RAEB2, based on the cantly deregulated gene pathways in early MDS are immuno- percentage of bone marrow blasts. -

Small Cell Ovarian Carcinoma: Genomic Stability and Responsiveness to Therapeutics
Gamwell et al. Orphanet Journal of Rare Diseases 2013, 8:33 http://www.ojrd.com/content/8/1/33 RESEARCH Open Access Small cell ovarian carcinoma: genomic stability and responsiveness to therapeutics Lisa F Gamwell1,2, Karen Gambaro3, Maria Merziotis2, Colleen Crane2, Suzanna L Arcand4, Valerie Bourada1,2, Christopher Davis2, Jeremy A Squire6, David G Huntsman7,8, Patricia N Tonin3,4,5 and Barbara C Vanderhyden1,2* Abstract Background: The biology of small cell ovarian carcinoma of the hypercalcemic type (SCCOHT), which is a rare and aggressive form of ovarian cancer, is poorly understood. Tumourigenicity, in vitro growth characteristics, genetic and genomic anomalies, and sensitivity to standard and novel chemotherapeutic treatments were investigated in the unique SCCOHT cell line, BIN-67, to provide further insight in the biology of this rare type of ovarian cancer. Method: The tumourigenic potential of BIN-67 cells was determined and the tumours formed in a xenograft model was compared to human SCCOHT. DNA sequencing, spectral karyotyping and high density SNP array analysis was performed. The sensitivity of the BIN-67 cells to standard chemotherapeutic agents and to vesicular stomatitis virus (VSV) and the JX-594 vaccinia virus was tested. Results: BIN-67 cells were capable of forming spheroids in hanging drop cultures. When xenografted into immunodeficient mice, BIN-67 cells developed into tumours that reflected the hypercalcemia and histology of human SCCOHT, notably intense expression of WT-1 and vimentin, and lack of expression of inhibin. Somatic mutations in TP53 and the most common activating mutations in KRAS and BRAF were not found in BIN-67 cells by DNA sequencing. -

Microglia Emerge from Erythromyeloid Precursors Via Pu.1- and Irf8-Dependent Pathways
ART ic LE S Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways Katrin Kierdorf1,2, Daniel Erny1, Tobias Goldmann1, Victor Sander1, Christian Schulz3,4, Elisa Gomez Perdiguero3,4, Peter Wieghofer1,2, Annette Heinrich5, Pia Riemke6, Christoph Hölscher7,8, Dominik N Müller9, Bruno Luckow10, Thomas Brocker11, Katharina Debowski12, Günter Fritz1, Ghislain Opdenakker13, Andreas Diefenbach14, Knut Biber5,15, Mathias Heikenwalder16, Frederic Geissmann3,4, Frank Rosenbauer6 & Marco Prinz1,17 Microglia are crucial for immune responses in the brain. Although their origin from the yolk sac has been recognized for some time, their precise precursors and the transcription program that is used are not known. We found that mouse microglia were derived from primitive c-kit+ erythromyeloid precursors that were detected in the yolk sac as early as 8 d post conception. + lo − + − + These precursors developed into CD45 c-kit CX3CR1 immature (A1) cells and matured into CD45 c-kit CX3CR1 (A2) cells, as evidenced by the downregulation of CD31 and concomitant upregulation of F4/80 and macrophage colony stimulating factor receptor (MCSF-R). Proliferating A2 cells became microglia and invaded the developing brain using specific matrix metalloproteinases. Notably, microgliogenesis was not only dependent on the transcription factor Pu.1 (also known as Sfpi), but also required Irf8, which was vital for the development of the A2 population, whereas Myb, Id2, Batf3 and Klf4 were not required. Our data provide cellular and molecular insights into the origin and development of microglia. Microglia are the tissue macrophages of the brain and scavenge dying have the ability to give rise to microglia and macrophages in vitro cells, pathogens and molecules using pattern recognition receptors and in vivo under defined conditions. -

Regulation of Cdc42 and Its Effectors in Epithelial Morphogenesis Franck Pichaud1,2,*, Rhian F
© 2019. Published by The Company of Biologists Ltd | Journal of Cell Science (2019) 132, jcs217869. doi:10.1242/jcs.217869 REVIEW SUBJECT COLLECTION: ADHESION Regulation of Cdc42 and its effectors in epithelial morphogenesis Franck Pichaud1,2,*, Rhian F. Walther1 and Francisca Nunes de Almeida1 ABSTRACT An overview of Cdc42 Cdc42 – a member of the small Rho GTPase family – regulates cell Cdc42 was discovered in yeast and belongs to a large family of small – polarity across organisms from yeast to humans. It is an essential (20 30 kDa) GTP-binding proteins (Adams et al., 1990; Johnson regulator of polarized morphogenesis in epithelial cells, through and Pringle, 1990). It is part of the Ras-homologous Rho subfamily coordination of apical membrane morphogenesis, lumen formation and of GTPases, of which there are 20 members in humans, including junction maturation. In parallel, work in yeast and Caenorhabditis elegans the RhoA and Rac GTPases, (Hall, 2012). Rho, Rac and Cdc42 has provided important clues as to how this molecular switch can homologues are found in all eukaryotes, except for plants, which do generate and regulate polarity through localized activation or inhibition, not have a clear homologue for Cdc42. Together, the function of and cytoskeleton regulation. Recent studies have revealed how Rho GTPases influences most, if not all, cellular processes. important and complex these regulations can be during epithelial In the early 1990s, seminal work from Alan Hall and his morphogenesis. This complexity is mirrored by the fact that Cdc42 can collaborators identified Rho, Rac and Cdc42 as main regulators of exert its function through many effector proteins. -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Anti-MTA2 Monoclonal Antibody, Clone NUB3-387 (DCABH-8980) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-MTA2 monoclonal antibody, clone NUB3-387 (DCABH-8980) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Product Overview Mouse monoclonal to MTA2 Antigen Description The p53 tumor suppressor gene integrates numerous signals that control cell life and death. There are several proteins that are involved in the p53 pathway, including Chk2, p53R2, p53AIP1, Noxa, PIDD, and MTA2. The transcriptional activity of p53 is modulated by protein stability and acetylation. MTA2, also termed MTA1L1, was found to be a subunit of nucleosome remodeling and deacetylating (NuRD) complex. MTA2 modulates the enzymatic activity of the histone deacetylase complex and its expression reduces the levels of acetylated p53. Deacetylation of p53 by MTA2 represses p53 dependent transcriptional activation and modulates p53 mediated cell growth arrest and apoptosis. MTA2 is ubiquitously expressed in human tissues. Immunogen Synthetic peptide: RRAARRPNLPLKVKPT conjugated to KLH, corresponding to amino acids 626-641 of Human MTA2 Isotype IgG1 Source/Host Mouse Species Reactivity Mouse, Rat, Hamster, Cow, Dog, Human, Monkey Clone NUB3-387 Purification This antibody is a purified IgG. Conjugate Unconjugated Applications ELISA, WB, IP, ICC/IF Positive Control ICC/IF: HEK-293T cells transfected with MTA2 expression vector WB: 293T total cell extract. Format Liquid Size 100 μl Buffer Preservative: 15mM Sodium Azide; Constituents: 10mM PBS, pH 7.4 Preservative 15mM Sodium Azide Storage Store at +4°C short term (1-2 weeks). Aliquot and store at -20°C or -80°C. Avoid repeated freeze 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved / thaw cycles. -

The Autophagy Receptor SQSTM1/P62 Mediates Anti-Inflammatory Actions of the Selective NR3C1/ Glucocorticoid Receptor Modulator Compound a (Cpda) in Macrophages
Autophagy ISSN: 1554-8627 (Print) 1554-8635 (Online) Journal homepage: http://www.tandfonline.com/loi/kaup20 The autophagy receptor SQSTM1/p62 mediates anti-inflammatory actions of the selective NR3C1/ glucocorticoid receptor modulator compound A (CpdA) in macrophages Viacheslav Mylka, Julie Deckers, Dariusz Ratman, Lode De Cauwer, Jonathan Thommis, Riet De Rycke, Francis Impens, Claude Libert, Jan Tavernier, Wim Vanden Berghe, Kris Gevaert & Karolien De Bosscher To cite this article: Viacheslav Mylka, Julie Deckers, Dariusz Ratman, Lode De Cauwer, Jonathan Thommis, Riet De Rycke, Francis Impens, Claude Libert, Jan Tavernier, Wim Vanden Berghe, Kris Gevaert & Karolien De Bosscher (2018) The autophagy receptor SQSTM1/p62 mediates anti- inflammatory actions of the selective NR3C1/glucocorticoid receptor modulator compound A (CpdA) in macrophages, Autophagy, 14:12, 2049-2064, DOI: 10.1080/15548627.2018.1495681 To link to this article: https://doi.org/10.1080/15548627.2018.1495681 © 2018 The Author(s). Published by Informa UK Limited, trading as Taylor & Francis Group. Published online: 14 Sep 2018. Submit your article to this journal Article views: 907 View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=kaup20 AUTOPHAGY 2018, VOL. 14, NO. 12, 2049–2064 https://doi.org/10.1080/15548627.2018.1495681 RESEARCH PAPER - BASIC SCIENCE The autophagy receptor SQSTM1/p62 mediates anti-inflammatory actions of the selective NR3C1/glucocorticoid receptor modulator -
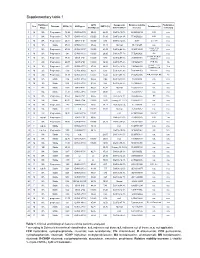
Supplementary Table 2 Supplementary Table 1
Supplementary table 1 Rai/ Binet IGHV Cytogenetic Relative viability Fludarabine- Sex Outcome CD38 (%) IGHV gene ZAP70 (%) Treatment (s) Stage identity (%) abnormalities* increase refractory 1 M 0/A Progressive 14,90 IGHV3-64*05 99,65 28,20 Del17p 18.0% 62,58322819 FCR n.a. 2 F 0/A Progressive 78,77 IGHV3-48*03 100,00 51,90 Del17p 24.8% 77,88052021 FCR n.a. 3 M 0/A Progressive 29,81 IGHV4-b*01 100,00 9,10 Del17p 12.0% 36,48 Len, Chl n.a. 4 M 1/A Stable 97,04 IGHV3-21*01 97,22 18,11 Normal 85,4191657 n.a. n.a. Chl+O, PCR, 5 F 0/A Progressive 87,00 IGHV4-39*07 100,00 43,20 Del13q 68.3% 35,23314039 n.a. HDMP+R 6 M 0/A Progressive 1,81 IGHV3-43*01 100,00 20,90 Del13q 77.7% 57,52490626 Chl n.a. Chl, FR, R-CHOP, 7 M 0/A Progressive 97,80 IGHV1-3*01 100,00 9,80 Del17p 88.5% 48,57389901 n.a. HDMP+R 8 F 2/B Progressive 69,07 IGHV5-a*03 100,00 16,50 Del17p 77.2% 107,9656878 FCR, BA No R-CHOP, FCR, 9 M 1/A Progressive 2,13 IGHV3-23*01 97,22 29,80 Del11q 16.3% 134,5866919 Yes Flavopiridol, BA 10 M 2/A Progressive 0,36 IGHV3-30*02 92,01 0,38 Del13q 81.9% 78,91844953 Unknown n.a. 11 M 2/B Progressive 15,17 IGHV3-20*01 100,00 13,20 Del11q 95.3% 75,52880995 FCR, R-CHOP, BR No 12 M 0/A Stable 0,14 IGHV3-30*02 90,62 7,40 Del13q 13.0% 13,0939004 n.a. -

Ten Commandments for a Good Scientist
Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Wageningen 2010 Thesis committee Thesis supervisors Prof. dr. ir. Ivonne M.C.M. Rietjens Professor of Toxicology Wageningen University Prof. dr. Albertinka J. Murk Personal chair at the sub-department of Toxicology Wageningen University Thesis co-supervisor Dr. ir. Jacques J.M. Vervoort Associate professor at the Laboratory of Biochemistry Wageningen University Other members Prof. dr. Michael R. Muller, Wageningen University Prof. dr. ir. Huub F.J. Savelkoul, Wageningen University Prof. dr. Everardus J. van Zoelen, Radboud University Nijmegen Dr. ir. Toine F.H. Bovee, RIKILT, Wageningen This research was conducted under the auspices of the Graduate School VLAG Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds Ana María Sotoca Covaleda Thesis submitted in fulfillment of the requirements for the degree of doctor at Wageningen University by the authority of the Rector Magnificus Prof. dr. M.J. Kropff, in the presence of the Thesis Committee appointed by the Academic Board to be defended in public on Tuesday 14 September 2010 at 4 p.m. in the Aula Unravelling the mechanism of differential biological responses induced by food-borne xeno- and phyto-estrogenic compounds. Ana María Sotoca Covaleda Thesis Wageningen University, Wageningen, The Netherlands, 2010, With references, and with summary in Dutch. ISBN: 978-90-8585-707-5 “Caminante no hay camino, se hace camino al andar. Al andar se hace camino, y al volver la vista atrás se ve la senda que nunca se ha de volver a pisar” - Antonio Machado – A mi madre. -

The UVB-Induced Gene Expression Profile of Human Epidermis in Vivo Is Different from That of Cultured Keratinocytes
Oncogene (2006) 25, 2601–2614 & 2006 Nature Publishing Group All rights reserved 0950-9232/06 $30.00 www.nature.com/onc ORIGINAL ARTICLE The UVB-induced gene expression profile of human epidermis in vivo is different from that of cultured keratinocytes CD Enk1, J Jacob-Hirsch2, H Gal3, I Verbovetski4, N Amariglio2, D Mevorach4, A Ingber1, D Givol3, G Rechavi2 and M Hochberg1 1Department of Dermatology, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel; 2Department of Pediatric Hemato-Oncology and Functional Genomics, Safra Children’s Hospital, Sheba Medical Center and Sackler School of Medicine, Tel-Aviv University,Tel Aviv, Israel; 3Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel and 4The Laboratory for Cellular and Molecular Immunology, Department of Medicine, The Hadassah-Hebrew University Medical Center, Jerusalem, Israel In order to obtain a comprehensive picture of the radiation. UVB, with a wavelength range between 290 molecular events regulating cutaneous photodamage of and 320 nm, represents one of the most important intact human epidermis, suction blister roofs obtained environmental hazards affectinghuman skin (Hahn after a single dose of in vivo ultraviolet (UV)B exposure and Weinberg, 2002). To protect itself against the were used for microarray profiling. We found a changed DNA-damaging effects of sunlight, the skin disposes expression of 619 genes. Half of the UVB-regulated genes over highly complicated cellular programs, including had returned to pre-exposure baseline levels at 72 h, cell-cycle arrest, DNA repair and apoptosis (Brash et al., underscoring the transient character of the molecular 1996). Failure in selected elements of these defensive cutaneous UVB response. -

Molecular Associations and Clinical Significance of Raps In
ORIGINAL RESEARCH published: 21 June 2021 doi: 10.3389/fmolb.2021.677979 Molecular Associations and Clinical Significance of RAPs in Hepatocellular Carcinoma Sarita Kumari 1†, Mohit Arora 2†, Jay Singh 1, Lokesh K. Kadian 2, Rajni Yadav 3, Shyam S. Chauhan 2* and Anita Chopra 1* 1Laboratory Oncology Unit, Dr. BRA-IRCH, All India Institute of Medical Sciences, New Delhi, India, 2Department of Biochemistry, All India Institute of Medical Sciences, New Delhi, India, 3Department of Pathology, All India Institute of Medical Sciences, New Delhi, India Hepatocellular carcinoma (HCC) is an aggressive gastrointestinal malignancy with a high rate of mortality. Multiple studies have individually recognized members of RAP gene family as critical regulators of tumor progression in several cancers, including hepatocellular carcinoma. These studies suffer numerous limitations including a small sample size and lack of analysis of various clinicopathological and molecular Edited by: Veronica Aran, features. In the current study, we utilized authoritative multi-omics databases to Instituto Estadual do Cérebro Paulo determine the association of RAP gene family expression and detailed molecular and Niemeyer (IECPN), Brazil clinicopathological features in hepatocellular carcinoma (HCC). All five RAP genes Reviewed by: were observed to harbor dysregulated expression in HCC compared to normal liver Pooja Panwalkar, Weill Cornell Medicine, United States tissues. RAP2A exhibited strongest ability to differentiate tumors from the normal Jasminka Omerovic, tissues. RAP2A expression was associated with progressive tumor grade, TP53 and University of Split, Croatia CTNNB1 mutation status. Additionally, RAP2A expression was associated with the *Correspondence: Anita Chopra alteration of its copy numbers and DNA methylation. RAP2A also emerged as an [email protected] independent marker for patient prognosis. -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement.