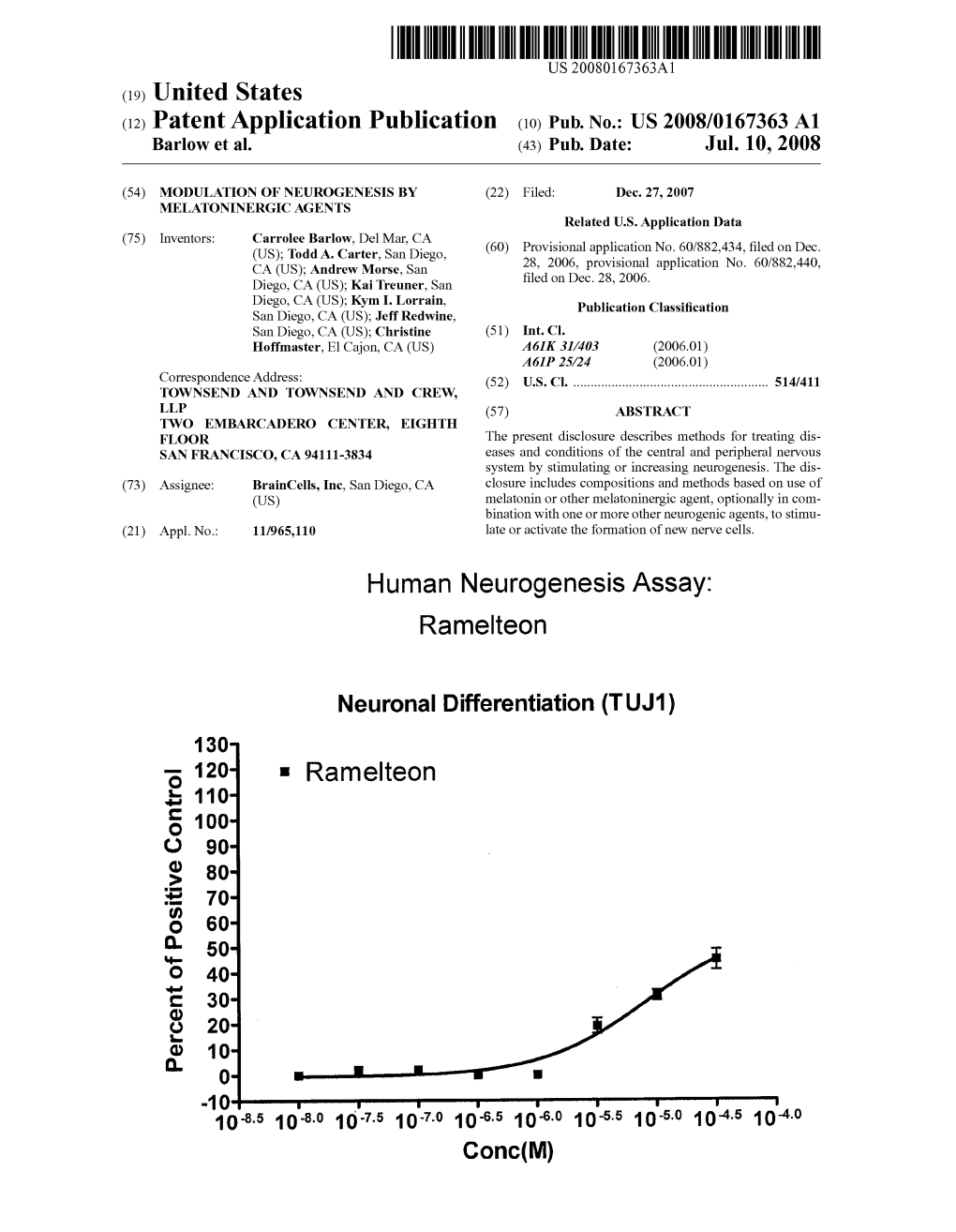
Human Neurogenesis Assay: Ramelteon
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

E30 SEM. O.C. Disclosed Is a Compound Represented by the Formula (1) (51) Int
USOO9453000B2 (12) United States Patent (10) Patent No.: US 9.453,000 B2 Kimura et al. (45) Date of Patent: *Sep. 27, 2016 (54) POLYCYCLIC COMPOUND (56) References Cited (75) Inventors: Teiji Kimura, Tsukuba (JP); Noritaka U.S. PATENT DOCUMENTS Kitazawa, Tsukuba (JP); Toshihiko 3,470,167 A 9, 1969 Sarkar Kaneko, Tsukuba (JP); Nobuaki Sato, 3,989,816 A 1 1/1976 Rajadhyaksha Tsukuba (JP); Koki Kawano, Tsukuba 4,910,200 A 3, 1990 Curtze et al. (JP): Koichi Ito, Tsukuba (JP); 5,281,626 A 1/1994 Oinuma et al. M s Tak ishi Tsukub JP 5,563,162 A 10, 1996 Oku et al. amoru Takaishi Tsukuba (JP); 5,804,577 A 9, 1998 Hebeisen et al. Takeo Sasaki, Tsukuba (JP); Yu 5,985,856 A 11/1999 Stella et al. Yoshida, Tsukuba (JP); Toshiyuki 6,235,728 B1 5, 2001 Golik et al. Uemura, Tsukuba (JP); Takashi Doko, g R 1939. E. al. Its SE E. Shinmyo, 7,138.414 B2 11/2006 Schoenafingereatch et al. et al. sukuba (JP); Daiju Hasegawa, 7,300,936 B2 11/2007 Parker et al. Tsukuba (JP); Takehiko Miyagawa, 7,314,940 B2 1/2008 Graczyk et al. Hatfield (GB); Hiroaki Hagiwara, 7,618,960 B2 11/2009 Kimura et al. Tsukuba (JP) 7,667,041 B2 2/2010 Kimura et al. 7,687,640 B2 3/2010 Kimura et al. 7,713,993 B2 5/2010 Kimura et al. (73) Assignee: EISAI R&D MANAGEMENT CO., 7,737,141 B2 6/2010 Kimura et al. LTD., Tokyo (JP) 7,880,009 B2 2/2011 Kimura et al. -

Nitrate Prodrugs Able to Release Nitric Oxide in a Controlled and Selective
Europäisches Patentamt *EP001336602A1* (19) European Patent Office Office européen des brevets (11) EP 1 336 602 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.7: C07C 205/00, A61K 31/00 20.08.2003 Bulletin 2003/34 (21) Application number: 02425075.5 (22) Date of filing: 13.02.2002 (84) Designated Contracting States: (71) Applicant: Scaramuzzino, Giovanni AT BE CH CY DE DK ES FI FR GB GR IE IT LI LU 20052 Monza (Milano) (IT) MC NL PT SE TR Designated Extension States: (72) Inventor: Scaramuzzino, Giovanni AL LT LV MK RO SI 20052 Monza (Milano) (IT) (54) Nitrate prodrugs able to release nitric oxide in a controlled and selective way and their use for prevention and treatment of inflammatory, ischemic and proliferative diseases (57) New pharmaceutical compounds of general effects and for this reason they are useful for the prep- formula (I): F-(X)q where q is an integer from 1 to 5, pref- aration of medicines for prevention and treatment of in- erably 1; -F is chosen among drugs described in the text, flammatory, ischemic, degenerative and proliferative -X is chosen among 4 groups -M, -T, -V and -Y as de- diseases of musculoskeletal, tegumental, respiratory, scribed in the text. gastrointestinal, genito-urinary and central nervous sys- The compounds of general formula (I) are nitrate tems. prodrugs which can release nitric oxide in vivo in a con- trolled and selective way and without hypotensive side EP 1 336 602 A1 Printed by Jouve, 75001 PARIS (FR) EP 1 336 602 A1 Description [0001] The present invention relates to new nitrate prodrugs which can release nitric oxide in vivo in a controlled and selective way and without the side effects typical of nitrate vasodilators drugs. -

Bifeprunox: a Novel Antipsychotic Agent with Partial Agonist Properties at Dopamine D2 and Serotonin 5-HT1A Receptors
DRUG EVALUATION Bifeprunox: a novel antipsychotic agent with partial agonist properties at dopamine D2 and serotonin 5-HT1A receptors Marie-Louise G Most second-generation, atypical, dopamine (DA) D2/5-HT2 blocking antipsychotics still Wadenberg induce extrapyramidal side effects (EPS) in higher doses. Weight gain and metabolic University of Kalmar, disturbances are also a problem, and negative and cognitive symptoms have not been Department of Natural Sciences, Norra Vagen 49, sufficiently addressed. The current brain DA mesolimbic hyperactive/mesocortical SE-391 82 Kalmar, Sweden hypoactive hypothesis of schizophrenia suggests that DA D2/5-HT1A receptor partial agonist Tel.: +46 480 446 277; properties may be more efficacious with less side effects. DA D2 receptor partial agonists Fax: +46 480 446 244; may stabilize a hyperactive/hypoactive DA condition. Additional 5-HT stimulation may marie-louise.wadenberg@ 1A hik.se enhance therapeutic efficacy and also improve EPS liability profile. In clinical trials in schizophrenic patients, the novel DA D2/5-HT1A partial agonist bifeprunox indeed demonstrates therapeutic efficacy, a safe EPS profile and appears beneficial regarding weight gain, prolactin, blood lipid and glucose levels and cardiac rhythm. The data on bifeprunox are promising and suggest that combined DA D2/5-HT1A partial agonism may well be important properties for future-generation antipsychotics. Bifeprunox, a novel antipsychotic agent with a (DA D1, D2, D4, 5-HT2A/C, 5-HT1A, hista- so-called third-generation atypical pharmacolog- mine H1, α1, α2, cholinergic muscarinic recep- ical profile, is currently in clinical trials and tor affinity) and comparatively lower affinity for expected to launch as a schizophrenia therapy in the DA D2 receptor than traditional APDs. -

HTR1A Polymorphisms and Clinical Efficacy Of
International Journal of Neuropsychopharmacology, (2016) 19(5): 1–10 doi:10.1093/ijnp/pyv125 Advance Access publication November 14, 2015 Research Article research article HTR1A Polymorphisms and Clinical Efficacy of Antipsychotic Drug Treatment in Schizophrenia: A Meta-Analysis Yoshiteru Takekita, MD, PhD; Chiara Fabbri, MD; Masaki Kato, MD, PhD; Yosuke Koshikawa, MA; Aran Tajika, MD; Toshihiko Kinoshita, MD, PhD; Alessandro Serretti, MD, PhD Department of Biomedical and NeuroMotor Sciences, University of Bologna, Bologna, Italy (Drs Takekita, Fabbri, and Serretti); Department of Neuropsychiatry, Kansai Medical University, Osaka, Japan (Drs Takekita, Kato, Koshikawa, and Kinoshita); Department of Health Promotion and Human Behavior, Kyoto University Graduate School of Medicine/School of Public Health, Kyoto, Japan (Dr Tajika). Correspondence: Yoshiteru Takekita, MD, PhD, Department of Biomedical and NeuroMotor Sciences, University of Bologna, Bologna, Viale Carlo Pepoli 5, Bologna, 40123, Italy ([email protected]). Abstract Background: This meta-analysis was conducted to evaluate whether HTR1A gene polymorphisms impact the efficacy of antipsychotic drugs in patients with schizophrenia. Methods: Candidate gene studies that were published in English up to August 6, 2015 were identified by a literature search of PubMed, Web of Science, and Google scholar. Data were pooled from individual clinical trials considering overall symptoms, positive symptoms and negative symptoms, and standard mean differences were calculated by applying a random-effects model. Results: The present meta-analysis included a total of 1281 patients from 10 studies. Three polymorphisms of HTR1A (rs6295, rs878567, and rs1423691) were selected for the analysis. In the pooled data from all studies, none of these HTR1A polymorphisms correlated significantly with either overall symptoms or positive symptoms. -

What Pigments Are in Plants?
BUILD YOUR FUTURE! ANYANG BEST COMPLETE MACHINERY ENGINEERING CO.,LTD WHAT PIGMENTS ARE IN PLANTS? Pigments Pigments are chemical compounds responsible for color in a range of living substances and in the inorganic world. Pigments absorb some of the light they receive, and so reflect only certain wavelengths of visible light. This makes them appear "colorful.” Cave paintings by early man show the early use of pigments, in a limited range from straw color to reddish brown and black. These colors occurred naturally in charcoals, and in mineral oxides such as chalk and ochre. The WebExhibit on Pigments has more information on these early painting palettes. Many early artists used natural pigments, but nowadays they have been replaced by cheaper and less toxic synthetic pigments. Biological Pigments Pigments are responsible for many of the beautiful colors we see in the plant world. Dyes have often been made from both animal sources and plant extracts . Some of the pigments found in animals have also recently been found in plants. Website: www.bestextractionmachine.com Email: [email protected] Tel: +86 372 5965148 Fax: +86 372 5951936 Mobile: ++86 8937276399 BUILD YOUR FUTURE! ANYANG BEST COMPLETE MACHINERY ENGINEERING CO.,LTD Major Plant Pigments White Bird Of Paradise Tree Bilirubin is responsible for the yellow color seen in jaundice sufferers and bruises, and is created when hemoglobin (the pigment that makes blood red) is broken down. Recently this pigment has also been found in plants, specifically in the orange fuzz on seeds of the white Bird of Paradise tree. The bilirubin in plants doesn’t come from breaking down hemoglobin. -

Treatment Protocol Copyright © 2018 Kostoff Et Al
Prevention and reversal of Alzheimer's disease: treatment protocol Copyright © 2018 Kostoff et al PREVENTION AND REVERSAL OF ALZHEIMER'S DISEASE: TREATMENT PROTOCOL by Ronald N. Kostoffa, Alan L. Porterb, Henry. A. Buchtelc (a) Research Affiliate, School of Public Policy, Georgia Institute of Technology, USA (b) Professor Emeritus, School of Public Policy, Georgia Institute of Technology, USA (c) Associate Professor, Department of Psychiatry, University of Michigan, USA KEYWORDS Alzheimer's Disease; Dementia; Text Mining; Literature-Based Discovery; Information Technology; Treatments Prevention and reversal of Alzheimer's disease: treatment protocol Copyright © 2018 Kostoff et al CITATION TO MONOGRAPH Kostoff RN, Porter AL, Buchtel HA. Prevention and reversal of Alzheimer's disease: treatment protocol. Georgia Institute of Technology. 2018. PDF. https://smartech.gatech.edu/handle/1853/59311 COPYRIGHT AND CREATIVE COMMONS LICENSE COPYRIGHT Copyright © 2018 by Ronald N. Kostoff, Alan L. Porter, Henry A. Buchtel Printed in the United States of America; First Printing, 2018 CREATIVE COMMONS LICENSE This work can be copied and redistributed in any medium or format provided that credit is given to the original author. For more details on the CC BY license, see: http://creativecommons.org/licenses/by/4.0/ This work is licensed under a Creative Commons Attribution 4.0 International License<http://creativecommons.org/licenses/by/4.0/>. DISCLAIMERS The views in this monograph are solely those of the authors, and do not represent the views of the Georgia Institute of Technology or the University of Michigan. This monograph is not intended as a substitute for the medical advice of physicians. The reader should regularly consult a physician in matters relating to his/her health and particularly with respect to any symptoms that may require diagnosis or medical attention. -

Mixed Antagonistic Effects of the Ginkgolides at Recombinant Human R1 GABAC Receptors
Neuropharmacology 63 (2012) 1127e1139 Contents lists available at SciVerse ScienceDirect Neuropharmacology journal homepage: www.elsevier.com/locate/neuropharm Mixed antagonistic effects of the ginkgolides at recombinant human r1 GABAC receptors Shelley H. Huang a, Trevor M. Lewis b, Sarah C.R. Lummis c, Andrew J. Thompson c, Mary Chebib d, Graham A.R. Johnston a, Rujee K. Duke a,* a Discipline of Pharmacology, School of Medical Sciences, Faculty of Medicine, University of Sydney, Australia b School of Medical Sciences, University of New South Wales, Australia c Department of Biochemistry, University of Cambridge, Cambridge, United Kingdom d Faculty of Pharmacy, University of Sydney, Australia article info abstract Article history: The diterpene lactones of Ginkgo biloba, ginkgolides A, B and C are antagonists at a range of Cys-loop Received 11 July 2011 receptors. This study examined the effects of the ginkgolides at recombinant human r1 GABAC recep- Received in revised form tors expressed in Xenopus oocytes using two-electrode voltage clamp. The ginkgolides were moderately 18 June 2012 potent antagonists with IC sinthemM range. At 10 mM, 30 mM and 100 mM, the ginkgolides caused Accepted 24 June 2012 50 rightward shifts of GABA doseeresponse curves and reduced maximal GABA responses, characteristic of noncompetitive antagonists, while the potencies showed a clear dependence on GABA concentration, Keywords: indicating apparent competitive antagonism. This suggests that the ginkgolides exert a mixed-type Ginkgolide Bilobalide antagonism at the r1 GABAC receptors. The ginkgolides did not exhibit any obvious use-dependent Mixed-antagonism inhibition. Fitting of the data to a number of kinetic schemes suggests an allosteric inhibition as Use-dependent a possible mechanism of action of the ginkgolides which accounts for their inhibition of the responses GABAr receptor without channel block or use-dependent inhibition. -

Prediction of Premature Termination Codon Suppressing Compounds for Treatment of Duchenne Muscular Dystrophy Using Machine Learning
Prediction of Premature Termination Codon Suppressing Compounds for Treatment of Duchenne Muscular Dystrophy using Machine Learning Kate Wang et al. Supplemental Table S1. Drugs selected by Pharmacophore-based, ML-based and DL- based search in the FDA-approved drugs database Pharmacophore WEKA TF 1-Palmitoyl-2-oleoyl-sn-glycero-3- 5-O-phosphono-alpha-D- (phospho-rac-(1-glycerol)) ribofuranosyl diphosphate Acarbose Amikacin Acetylcarnitine Acetarsol Arbutamine Acetylcholine Adenosine Aldehydo-N-Acetyl-D- Benserazide Acyclovir Glucosamine Bisoprolol Adefovir dipivoxil Alendronic acid Brivudine Alfentanil Alginic acid Cefamandole Alitretinoin alpha-Arbutin Cefdinir Azithromycin Amikacin Cefixime Balsalazide Amiloride Cefonicid Bethanechol Arbutin Ceforanide Bicalutamide Ascorbic acid calcium salt Cefotetan Calcium glubionate Auranofin Ceftibuten Cangrelor Azacitidine Ceftolozane Capecitabine Benserazide Cerivastatin Carbamoylcholine Besifloxacin Chlortetracycline Carisoprodol beta-L-fructofuranose Cilastatin Chlorobutanol Bictegravir Citicoline Cidofovir Bismuth subgallate Cladribine Clodronic acid Bleomycin Clarithromycin Colistimethate Bortezomib Clindamycin Cyclandelate Bromotheophylline Clofarabine Dexpanthenol Calcium threonate Cromoglicic acid Edoxudine Capecitabine Demeclocycline Elbasvir Capreomycin Diaminopropanol tetraacetic acid Erdosteine Carbidopa Diazolidinylurea Ethchlorvynol Carbocisteine Dibekacin Ethinamate Carboplatin Dinoprostone Famotidine Cefotetan Dipyridamole Fidaxomicin Chlormerodrin Doripenem Flavin adenine dinucleotide -

)&F1y3x PHARMACEUTICAL APPENDIX to THE
)&f1y3X PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE )&f1y3X PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 3 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. Product CAS No. Product CAS No. ABAMECTIN 65195-55-3 ACTODIGIN 36983-69-4 ABANOQUIL 90402-40-7 ADAFENOXATE 82168-26-1 ABCIXIMAB 143653-53-6 ADAMEXINE 54785-02-3 ABECARNIL 111841-85-1 ADAPALENE 106685-40-9 ABITESARTAN 137882-98-5 ADAPROLOL 101479-70-3 ABLUKAST 96566-25-5 ADATANSERIN 127266-56-2 ABUNIDAZOLE 91017-58-2 ADEFOVIR 106941-25-7 ACADESINE 2627-69-2 ADELMIDROL 1675-66-7 ACAMPROSATE 77337-76-9 ADEMETIONINE 17176-17-9 ACAPRAZINE 55485-20-6 ADENOSINE PHOSPHATE 61-19-8 ACARBOSE 56180-94-0 ADIBENDAN 100510-33-6 ACEBROCHOL 514-50-1 ADICILLIN 525-94-0 ACEBURIC ACID 26976-72-7 ADIMOLOL 78459-19-5 ACEBUTOLOL 37517-30-9 ADINAZOLAM 37115-32-5 ACECAINIDE 32795-44-1 ADIPHENINE 64-95-9 ACECARBROMAL 77-66-7 ADIPIODONE 606-17-7 ACECLIDINE 827-61-2 ADITEREN 56066-19-4 ACECLOFENAC 89796-99-6 ADITOPRIM 56066-63-8 ACEDAPSONE 77-46-3 ADOSOPINE 88124-26-9 ACEDIASULFONE SODIUM 127-60-6 ADOZELESIN 110314-48-2 ACEDOBEN 556-08-1 ADRAFINIL 63547-13-7 ACEFLURANOL 80595-73-9 ADRENALONE -

PHARMACEUTICAL APPENDIX to the TARIFF SCHEDULE 2 Table 1
Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2020) Revision 19 Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names INN which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service CAS registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. -

A Synthetic Overview of New Molecules with 5-HT1A Binding Affinities
77 A Synthetic Overview of New Molecules with 5-HT1A Binding Affinities Hernán Pessoa-Mahana* 1 ; Ramiro Araya-Maturana1 , Claudio Saitz, B.1 and C. David Pessoa-Mahana2 1Departamento de Química Orgánica y Fisicoquímica. Facultad de Ciencias Químicas y Farmacéuticas. Universidad de Chile. Olivos 1007.Casilla 233. Santiago 1. Chile 2Departamento de Farmacia. Facultad de Química. Pontificia Universidad Católica de Chile. Vicuña Mackenna 4860-Casilla 306, Correo 22 Santiago-Chile Abstract: The present review discusses the synthetic strategies of new ligands exhibiting mainly 5-HT1A binding affinities. Specifically we focused our attention in the synthesis of compounds structurally related to arylpiperazine, 2-aminotetralin, and benzopyran derivatives. Keywords: serotonin, 5-HT1A ligands, arylpiperazines, aminotetralins, benzopyrans. INTRODUCTION During the last 15 years, seven distinct families of 5-HT receptors have been identified (5-HT1–5-HT7), and at least Depression is one of the most common illnesses, 15 subpopulations have been described for several of these affecting up to one-third of all people at the same time. [4,5]. The 5-HT1A receptors represent a major target for Depressive disorders encompass a variety of conditions neurobiological research and drug developments. A study on including two major forms of unipolar depression (i.e. major distribution of 5-HT1A receptors in the brains of various depression and dysthymia), adjustment disorders, animal species indicates that the highest densities are located subsyndromal depression (minor depression), seasonal in the hippocampus, septum, amygdale, and cortical limbic affective disorder (SAD), premenstrual dysphoric disorder areas. The 5-HT1A receptors located in the raphe nuclei are (PMDD), postpartum depression, atypical depression and known as somatodendritic autoreceptors. -

5-HT3 Receptor Antagonists in Neurologic and Neuropsychiatric Disorders: the Iceberg Still Lies Beneath the Surface
1521-0081/71/3/383–412$35.00 https://doi.org/10.1124/pr.118.015487 PHARMACOLOGICAL REVIEWS Pharmacol Rev 71:383–412, July 2019 Copyright © 2019 by The Author(s) This is an open access article distributed under the CC BY-NC Attribution 4.0 International license. ASSOCIATE EDITOR: JEFFREY M. WITKIN 5-HT3 Receptor Antagonists in Neurologic and Neuropsychiatric Disorders: The Iceberg Still Lies beneath the Surface Gohar Fakhfouri,1 Reza Rahimian,1 Jonas Dyhrfjeld-Johnsen, Mohammad Reza Zirak, and Jean-Martin Beaulieu Department of Psychiatry and Neuroscience, Faculty of Medicine, CERVO Brain Research Centre, Laval University, Quebec, Quebec, Canada (G.F., R.R.); Sensorion SA, Montpellier, France (J.D.-J.); Department of Pharmacodynamics and Toxicology, School of Pharmacy, Mashhad University of Medical Sciences, Mashhad, Iran (M.R.Z.); and Department of Pharmacology and Toxicology, University of Toronto, Toronto, Ontario, Canada (J.-M.B.) Abstract. ....................................................................................384 I. Introduction. ..............................................................................384 II. 5-HT3 Receptor Structure, Distribution, and Ligands.........................................384 A. 5-HT3 Receptor Agonists .................................................................385 B. 5-HT3 Receptor Antagonists. ............................................................385 Downloaded from 1. 5-HT3 Receptor Competitive Antagonists..............................................385 2. 5-HT3 Receptor