Chapter 11 - Alcohols and Ethers1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 21 the Chemistry of Carboxylic Acid Derivatives
Instructor Supplemental Solutions to Problems © 2010 Roberts and Company Publishers Chapter 21 The Chemistry of Carboxylic Acid Derivatives Solutions to In-Text Problems 21.1 (b) (d) (e) (h) 21.2 (a) butanenitrile (common: butyronitrile) (c) isopentyl 3-methylbutanoate (common: isoamyl isovalerate) The isoamyl group is the same as an isopentyl or 3-methylbutyl group: (d) N,N-dimethylbenzamide 21.3 The E and Z conformations of N-acetylproline: 21.5 As shown by the data above the problem, a carboxylic acid has a higher boiling point than an ester because it can both donate and accept hydrogen bonds within its liquid state; hydrogen bonding does not occur in the ester. Consequently, pentanoic acid (valeric acid) has a higher boiling point than methyl butanoate. Here are the actual data: INSTRUCTOR SUPPLEMENTAL SOLUTIONS TO PROBLEMS • CHAPTER 21 2 21.7 (a) The carbonyl absorption of the ester occurs at higher frequency, and only the carboxylic acid has the characteristic strong, broad O—H stretching absorption in 2400–3600 cm–1 region. (d) In N-methylpropanamide, the N-methyl group is a doublet at about d 3. N-Ethylacetamide has no doublet resonances. In N-methylpropanamide, the a-protons are a quartet near d 2.5. In N-ethylacetamide, the a- protons are a singlet at d 2. The NMR spectrum of N-methylpropanamide has no singlets. 21.9 (a) The first ester is more basic because its conjugate acid is stabilized not only by resonance interaction with the ester oxygen, but also by resonance interaction with the double bond; that is, the conjugate acid of the first ester has one more important resonance structure than the conjugate acid of the second. -

Recent Advances in Titanium Radical Redox Catalysis
JOCSynopsis Cite This: J. Org. Chem. 2019, 84, 14369−14380 pubs.acs.org/joc Recent Advances in Titanium Radical Redox Catalysis Terry McCallum, Xiangyu Wu, and Song Lin* Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States ABSTRACT: New catalytic strategies that leverage single-electron redox events have provided chemists with useful tools for solving synthetic problems. In this context, Ti offers opportunities that are complementary to late transition metals for reaction discovery. Following foundational work on epoxide reductive functionalization, recent methodological advances have significantly expanded the repertoire of Ti radical chemistry. This Synopsis summarizes recent developments in the burgeoning area of Ti radical catalysis with a focus on innovative catalytic strategies such as radical redox-relay and dual catalysis. 1. INTRODUCTION a green chemistry perspective, the abundance and low toxicity of Ti make its complexes highly attractive as reagents and Radical-based chemistry has long been a cornerstone of 5 1 catalysts in organic synthesis. synthetic organic chemistry. The high reactivity of organic IV/III radicals has made possible myriad new reactions that cannot be A classic example of Ti -mediated reactivity is the reductive ring opening of epoxides. This process preferentially readily achieved using two-electron chemistry. However, the − high reactivity of organic radicals is a double-edged sword, as cleaves and functionalizes the more substituted C O bond, the selectivity of these fleeting intermediates can be difficult to providing complementary regioselectivity to Lewis acid control in the presence of multiple chemotypes. In addition, promoted epoxide reactions. The synthetic value of Ti redox catalysis has been highlighted by their many uses in total catalyst-controlled regio- and stereoselective reactions involv- 6−10 ing free-radical intermediates remain limited,2 and the synthesis (Scheme 1). -

1.1 10 Oxidation of Alcohols and Aldehydes
1.1 10 Oxidation of alcohols and aldehydes By the end of this spread, you should be able to … 1Describe the oxidation of primary alcohols to form aldehydes and carboxylic acids. 1Describe the oxidation of secondary alcohols to form ketones. 1Describe the oxidation of aldehydes to form carboxylic acids. Key definition Oxidation of alcohols You will recall from your AS chemistry studies that primary and secondary alcohols can A redox reaction is one in which both reduction and oxidation take place. be oxidised using an oxidising agent. s !SUITABLEOXIDISINGAGENTISASOLUTIONCONTAININGACIDIlEDDICHROMATEIONS + 2− H /Cr2O7 . s 4HEOXIDISINGMIXTURECANBEMADEFROMPOTASSIUMDICHROMATE +2Cr2O7, and sulfuric acid, H2SO. During the reaction, the acidified potassium dichromate changes from orange to green. Remember that tertiary alcohols are not oxidised by acidified dichromate ions. Primary alcohols Primary alcohols can be oxidised to aldehydes and to carboxylic acids (see AS Chemistry spread 2.2.3). H H H O H O Oxidation Oxidation H CCOH H CC H CC H H H H H OH Primary alcohol Aldehyde Carboxylic acid Figure 1 Ethanol oxidised to ethanal, and finally to ethanoic acid The equation for the oxidation of ethanol to the aldehyde ethanal is shown below. Note that the oxidising agent is shown as [O] – this simplifies the equation. CH3CH2OH + [O] }m CH3CHO + H2O When preparing aldehydes in the laboratory, you will need to distil the aldehyde from the reaction mixture as it is formed. This prevents the aldehyde from being oxidised further to a carboxylic acid. Key definition When making the carboxylic acid, the reaction mixture is usually heated under reflux before distilling the product off. -

Chemistry 0310 - Organic Chemistry 1 Chapter 6
Dr. Peter Wipf Chemistry 0310 - Organic Chemistry 1 Chapter 6. SN2 Reactions Nucleophilic substitution reactions occur when nucleophiles (electron-rich species) displace leaving groups on electrophiles (electron-poor species). The net result is the substitution of one group for another bonded to a carbon atom. Nucleophilicity is increased by a negative charge and an increase in the polarizability. The greater the size and the lower the electronegativity of an atom, the greater its polarizability. Leaving groups are stable species that can be detached with their bonding electrons from a molecule during reaction. Most good leaving groups are the conjugate bases of strong acids. Sulfonates (mesylates, tosylates, triflates, etc.) are popular leaving groups since they can be readily obtained from alcohols. Solvents also influence nucleophilicity. SN2 reactions are second-order reactions whose rates depend o the concentration of both the alkyl halide and the nucleophile. The transition state of the one-step SN2 process involves a trigonal bipyramidal carbon and results in an overall inversion of configuration. The order of reactivity of alkyl halides and alkyl sulfonates in SN2 reactions is CH3>1°>2°>3°. The activation energy, DG‡, determines the rate of a reaction: k = koexp(-DG‡/RT). The overall change in free energy, DG, determines the position of the thermodynamic equilibrium. A positive sign of DG is characteristic for an endergonic reaction, a negative DG is found for an exergonic process. The rate of a reaction can be measured and provides information about its mechanism. The reaction order is the sum of the exponents in the rate equation. Relative Leaving Group Abilities: Leaving Group Common Name krel AcO acetate 1 x 10-10 Cl chloride 0.0001 Br bromide 0.001 I iodide 0.01 O mesylate 1.00 H3C S O O O tosylate 0.70 H3C S O O O brosylate 2.62 Br S O O O nosylate 13 O2N S O O O fluorosulfate 29,000 F S O O O triflate 56,000 CF3 S O O O nonaflate 120,000 C4F9 S O O. -

United States Patent 15) 3,669,956 Borck Et Al
United States Patent 15) 3,669,956 Borck et al. (45) June 13, 1972 54) 4-SUBSTITUTEDAMNO 260/472, 260/516, 260/518 R, 260/518 A, 260/519, PHENYACETIC ACDS AND 260/556 AR, 260/556 B, 260/558 S, 260/558 A, DERVATIVES THEREOF 260/559 T, 260/559 A, 260/.571, 260/574, 260/.575, (72 Inventors: Joachim Borck; Johann Dahin; Volker 424/244, 424/246, 424/248, 424/250, 424/267, Koppe; Josef Kramer; Gustav Shorre; J. 424/270, 424/272, 424/273, 424/274, 424/304, W. Hermann Hovy; Ernst Schorscher, all 424/309, 424/32 i, 424/324, 424/330 of Darmstadt, Germany 51) int. Cl. ........................................................ C07d 41/04 58) Field of Search........ 260/294X,293.4, 293.47, 239 BF, 73) Assignee: E. Merck A. G., Darmstadt, Germany 260/326.3, 294.3 E (22) Filed: July 22, 1968 56) References Cited (21) Appl. No.: 746,326 UNITED STATES PATENTS (30) Foreign Application Priority Data 3,252,970 5/1966 Huebner................................ 260/239 July 22, 1967 Germany.............................. M 74881 3,385,852 5/1968 Casadio................................. 260/246 Jan. 8, 1968 Germany... ....M 76850 OTHER PUBLICATIONS Feb. 23, 1968 Germany...... ...M 77363 March 1, 1968 Germany.............................. M 77429 Norman et al., J. Chen. Soc. 1963, (Nov.), 5431-6. (52) U.S. Cl................... 260/239 BF, 260/239 A, 260/239 E, Primary Examiner-Henry R. Jiles 260/243 B, 260/246, 260/247. 1, 260/247.2 R, Assistant Examiner-G. Thomas Todd 260/247.2 A, 260/247.2 B, 260/247.5 R, 260/247.7 Attorney-Millen, Raptes & White A, 260/247.7 H, -

Functionalized Hybrid Silicones – Catalysis, Synthesis and Application
Technische Universität München Fakultät für Chemie Fachgebiet Molekulare Katalyse Functionalized Hybrid Silicones – Catalysis, Synthesis and Application Sophie Luise Miriam Putzien Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. M. Schuster Prüfer der Dissertation: 1. Univ.-Prof. Dr. F. E. Kühn 2. Univ.-Prof. Dr. O.Nuyken (i.R.) Die Dissertation wurde am 16.02.2012 bei der Technischen Universität München eingereicht und durch die Fakultät für Chemie am 08.03.2012 angenommen. The following dissertation was prepared between April 2009 and March 2012 at the Chair of Inorganic Chemistry, Department of Molecular Catalysis of the Technische Universität München. I would like to express my deep gratitude to my academic supervisor Prof. Dr. Fritz E. Kühn for his support and confidence and the freedom of scientific research. This work was supported by a research grant from the BASF Construction Chemicals GmbH, Trostberg, Germany. Acknowledgement I would like to express my sincere gratitude to Prof. Dr. Oskar Nuyken and Dr. Eckhart Louis for their ongoing support and their undamped enthusiasm for my research topic. They supported this work with many inspiring discussions, new ideas and critical questions. I thank the BASF Construction Chemicals GmbH, Trostberg, for giving me the opportunity to work on an industrial cooperation project. Especially, I would like to thank Dr. Simone Klapdohr and Dr. Burkhard Walther, who accompanied this project from the industrial perspectice, for their support and the nice time I had in Trostberg during the application technological tests. -

Unusual Regioselectivity in the Opening of Epoxides by Carboxylic Acid Enediolates
Molecules 2008, 13, 1303-1311 manuscripts; DOI: 10.3390/molecules13061303 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.org/molecules Communication Unusual Regioselectivity in the Opening of Epoxides by Carboxylic Acid Enediolates Luis R. Domingo, Salvador Gil, Margarita Parra* and José Segura Department of Organic Chemistry, Universitat de València, Dr. Moliner 50, 46100 Burjassot, Spain. Fax +34(9)63543831; E-mails: [email protected]; [email protected]; [email protected] * Author to whom correspondence should be addressed; E-mail: [email protected] Received: 29 May 2008; in revised form: 5 June 2008 / Accepted: 6 June 2008 / Published: 9 June 2008 Abstract: Addition of carboxylic acid dianions appears to be a potential alternative to the use of aluminium enolates for nucleophilic ring opening of epoxides. These conditions require the use of a sub-stoichiometric amount of amine (10% mol) for dianion generation and the previous activation of the epoxide with LiCl. Yields are good, with high regioselectivity, but the use of styrene oxide led, unexpectedly, to a mixture resulting from the attack on both the primary and secondary carbon atoms. Generally, a low diastereoselectivity is seen on attack at the primary center, however only one diastereoisomer was obtained from attack to the secondary carbon of the styrene oxide. Keywords: Lactones, lithium chloride, nucleophilic addition, regioselectivity, diastereoselectivity. Introduction Epoxides have been recognized among the most versatile compounds in organic synthesis, not only as final products [1] but as key intermediates for further manipulations. Accordingly, new synthetic developments are continuously being published [2]. Due to its high ring strain (around 27 Kcal/mol) its ring-opening, particularly with carbon-based nucleophiles, is a highly valuable synthetic strategy Molecules 2008, 13 1304 [3]. -

Highly Efficient Epoxidation of Olefins with Hydrogen Peroxide Oxidant Using Modified Silver Polyoxometalate Catalysts
African Journal of Pure and Applied Chemistry Vol. 7(2), pp. 50-55, February 2013 Available online at http://www.academicjournals.org/AJPAC DOI: 10.5897/AJPAC12.060 ISSN 1996 - 0840 ©2013 Academic Journals Full Length Research Paper Highly efficient epoxidation of olefins with hydrogen peroxide oxidant using modified silver polyoxometalate catalysts Emmanuel Tebandeke 1*, Henry Ssekaalo 1 and Ola F. Wendt 2 1Department of Chemistry, Makerere University, P. O. Box 7062 Kampala, Uganda. 2Organic Chemistry, Department of Chemistry, Lund University, P. O. Box 124, 221 00 Lund, Sweden. Accepted 31 January, 2013 The catalytic epoxidation of olefins is an important reaction in chemical industry since epoxides are versatile and important intermediates in the synthesis of fine chemicals and pharmaceuticals. The current epoxidation procedures often use stoichiometric organic and sometimes toxic inorganic oxidants that are less favourable from an environmental and economical point of view. In contrast to such processes, catalytic epoxidation processes employing H2O2 oxidant are preferred for green processing. Herein is reported a highly efficient green process for the epoxidation of olefins using H2O2 oxidant and modified silver polyoxometalate catalysts at 65°C. The preparation and activity of the catalysts are described. The method enjoys >90% conversion and ≥99% selectivity to the epoxide for a variety of cyclic and linear olefins including terminal ones. The catalysts are easily recovered by filtration and are reusable several times. Key words: Epoxidation, olefins, hydrogen peroxide, polyoxometalate, silver catalysts. INTRODUCTION The catalytic epoxidation of olefins is very important in compared to organic peroxides and peracids (Jones, the chemical industry since epoxides are versatile and 1999; Lane and Burgess, 2003). -

Part I. the Total Synthesis Of
AN ABSTRACT OF THE THESIS OF Lester Percy Joseph Burton forthe degree of Doctor of Philosophy in Chemistry presentedon March 20, 1981. Title: Part 1 - The Total Synthesis of (±)-Cinnamodialand Related Drimane Sesquiterpenes Part 2 - Photochemical Activation ofthe Carboxyl Group Via NAcy1-2-thionothiazolidines Abstract approved: Redacted for privacy DT. James D. White Part I A total synthesis of the insect antifeedant(±)-cinnamodial ( ) and of the related drimanesesquiterpenes (±)-isodrimenin (67) and (±)-fragrolide (72)are described from the diene diester 49. Hydro- boration of 49 provided the C-6oxygenation and the trans ring junction in the form of alcohol 61. To confirm the stereoselectivity of the hydroboration, 61 was convertedto both (t)-isodrimenin (67) and (±)-fragrolide (72) in 3 steps. A diisobutylaluminum hydride reduction of 61 followed by a pyridiniumchlorochromate oxidation and treatment with lead tetraacetate yielded the dihydrodiacetoxyfuran102. The base induced elimination of acetic acid preceded theepoxidation and provided 106 which contains the desired hydroxy dialdehydefunctionality of cinnamodial in a protected form. The epoxide 106 was opened with methanol to yield the acetal 112. Reduction, hydrolysis and acetylation of 112 yielded (t)- cinnamodial in 19% overall yield. Part II - Various N- acyl- 2- thionothiazolidineswere prepared and photo- lysed in the presence of ethanol to provide the corresponding ethyl esters. The photochemical activation of the carboxyl function via these derivatives appears, for practical purposes, to be restricted tocases where a-keto hydrogen abstraction and subsequent ketene formation is favored by acyl substitution. Part 1 The Total Synthesis of (±)-Cinnamodial and Related Drimane Sesquiterpenes. Part 2 Photochemical Activation of the Carboxyl Group via N-Acy1-2-thionothiazolidines. -
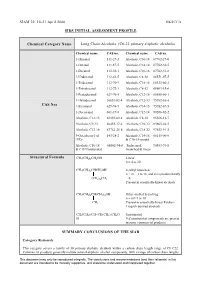
Long Chain Alcohols (C6-22 Primary Aliphatic Alcohols)
SIAM 22, 18-21 April 2006 UK/ICCA SIDS INITIAL ASSESSMENT PROFILE Chemical Category Name Long Chain Alcohols (C6-22 primary aliphatic alcohols) Chemical name CAS no. Chemical name CAS no. 1-Hexanol 111-27-3 Alcohols, C16-18 67762-27-0 1-Octanol 111-87-5 Alcohols, C14-18 67762-30-5 1-Decanol 112-30-1 Alcohols, C10-16 67762-41-8 1-Undecanol 112-42-5 Alcohols, C8-18 68551-07-5 1-Tridecanol 112-70-9 Alcohols, C14-16 68333-80-2 1-Tetradecanol 112-72-1 Alcohols, C6-12 68603-15-6 1-Pentadecanol 629-76-5 Alcohols, C12-16 68855-56-1 1-Hexadecanol 36653-82-4 Alcohols, C12-13 75782-86-4 CAS Nos 1-Eicosanol 629-96-9 Alcohols, C14-15 75782-87-5 1-Docosanol 661-19-8 Alcohols, C12-14 80206-82-2 Alcohols, C12-15 63393-82-8 Alcohols, C8-10 85566-12-7 Alcohols, C9-11 66455-17-2 Alcohols, C10-12 85665-26-5 Alcohols, C12-18 67762-25-8 Alcohols, C18-22 97552-91-5 9-Octadecen-1-ol 143-28-2 Alcohols, C14-18. 68155-00-0 (9Z)- & C16-18-unsatd Alcohols, C16-18 68002-94-8 Tridecanol, 90583-91-8 & C18 Unsaturated branched & linear Structural Formula CH3(CH2)nCH2OH Linear n = 4 to 20 CH3(CH2)nCHCH2OH 2-Alkyl branched n + m = 3 to 18, and m is predominantly (CH2)mCH3 = 0. Present in essentially-linear alcohols CH3(CH2)nCH(CH2)mOH Other-methyl branching n + m= 9 or 10 CH3 Present in essentially-linear Fischer- Tropsch derived alcohols CH3(CH2)7CH=CH(CH2)7CH2O Unsaturated H 9-Z unsaturated components are present in some commercial products. -

Leaving Group of Substrate Important
1 Recap... • Last two lectures we looked at substitution reactions • We found that there we two (extreme) mechanisms • SN1 H H H H H PhS R H R Br R SPh Br • And SN2 H H H H PhS + Br R Br PhS R • We looked at many of the factors that influenced which ..mechanism was operating... • Now we will turn our attention to elimination reactions 2 Elimination reactions NaOH NaOH or low Br H2O OH concentration • You have seen SN1 substitution • Reaction not sped up by altering nucleophile - it is not in RDS • In fact if we increase the amount / concentration of nucleophile we get the following... high O H + + + Br Br OH concentration H H • We observe an elimination reaction • Overall HBr is lost from the molecule • Isn't chemistry fun - these are the same conditions as substitution! • Next two lectures will look at the mechanisms for elimination • And how we control the nature of the reaction we observe... 3 Elimination, bimolecular E2 O H Br HO Br H H • E2 - Elimination bimolecular or 2nd order reaction • 2 molecules in the RDS or transition state H H H H H C H C H H C Br Br H O H C H O H C C H H H H C H C H H H • Two molecules in the rate determining step • So both the base and the substrate control the reaction • Both carbon skeleton & leaving group of substrate important In substitutions nucleophile acts as a nucleophile & attacks carbon In eliminations nucleophile acts as a base & attacks proton 4 Elimination, bimolecular E2: MO • Little confusing as need bonding & anti-bonding in same diagram! • Orbitals need to be parallel for maximum overlap -

Carbonyl Chemistry I: Additions to Carbonyl Groups
Paul Bracher Chem 30 – Section 4 Carbonyl Chemistry I: Additions to Carbonyl Groups Section Agenda 1) Lessons from Exam I, Course Feedbeck Mechanism on Web Site 2) Carbonyl Groups: Structure, Bonding, Molecular Orbitals, Geometry, Reactivity 3) Handout: In-Section Problem Set 4) Handout: In-Section Solution Set 5) Weekly Office Hours: Thursday, 3-4PM and 8-9PM, in Bauer Lobby Structure and Bonding · Carbonyl carbons are roughly sp2 hybridized Molecular Orbital Picture and planar with bond angles near 120 degrees bonding p orbital antibonding p* orbital · The C=O p bond is formed by the overlap of two unhybridized p orbitals. The bond is polarized due to the different electronegativities of carbon and oxygen. 105 o Resonance Model O O Bürgi-Dunitz angle · Another way to picture carbonyl groups is I II through an MO picture. Since the coefficient of · Resonance form II suggests that the carbon pO’s contribution is greater in pC=O, then atom is surrounded by less negative charge conservation of blending atomic orbitals into density than oxygen, rendering the carbon more molecular orbitals tells you that pC must be a electrophilic. larger contributor to the p*C=O orbital. · When reagents add to carbonyl double bonds, · The larger coefficient on oxygen in the filled the nucleophile attacks the carbon atom. orbital explains why the oxygen atom behaves as a Lewis base (it commonly complexes with · Due to the increased negative charge density Lewis acids). The larger coefficient on carbon on oxygen, Lewis acids (such as protons) in the empty antibonding orbital explains why it commonly complex with oxygen.