SV&SNP A3.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
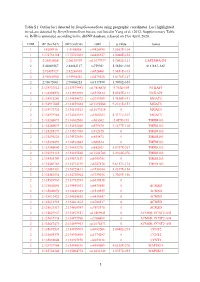
1 Table S1. Outlier Loci Detected by Deepgenomescan Using
Table S1. Outlier loci detected by DeepGenomeScan using geographic coordinates. Loci highlighted in red are detected by DeepGenomeScan but are not listed in Yang et al. (2012; Supplementary Table 4). RsID is annotated according to the dbSNP database released on 21st April, 2020. CHR BP (Grch37) BP (Grch38) rsID p.value Genes 1 1:4208918 1:4148858 rs9426495 3.8582E-104 1 1:175738168 1:175769032 rs6425357 2.0088E-105 2 2:20310668 2:20110907 rs11679737 1.2002E-111 LAPTM4A-DT 2 2:40289267 2:40062127 rs759361 5.5658E-104 SLC8A1-AS1 2 2:82495127 2:82268003 rs6726401 3.9451E-103 2 2:96660300 2:95994552 rs2579520 1.8176E-127 2 2:96672001 2:96006253 rs1917890 3.7698E-104 2 2:134333012 2:133575441 rs17816830 9.702E-105 NCKAP5 2 2:134350570 2:133592999 rs6715224 5.8745E-111 NCKAP5 2 2:134912243 2:134154672 rs2139309 1.7418E-191 MGAT5 2 2:134917005 2:134159434 rs11692586 9.2121E-151 MGAT5 2 2:134972732 2:134215161 rs11679218 0 MGAT5 2 2:134979966 2:134222395 rs1965183 5.3171E-107 MGAT5 2 2:135260071 2:134502500 rs503562 2.057E-153 TMEM163 2 2:135280039 2:134522468 rs579670 3.1477E-118 TMEM163 2 2:135285279 2:134527708 rs512375 0 TMEM163 2 2:135290221 2:134532650 rs655472 0 TMEM163 2 2:135290453 2:134532882 rs666614 0 TMEM163 2 2:135340840 2:134583270 rs842361 5.6717E-237 TMEM163 2 2:135393110 2:134635540 rs11684785 2.2164E-276 TMEM163 2 2:135430709 2:134673139 rs6745983 0 TMEM163 2 2:135469769 2:134712199 rs6747870 9.6117E-114 TMEM163 2 2:135483381 2:134725811 rs3739034 5.0357E-154 2 2:135483534 2:134725964 rs3739036 3.7269E-156 2 2:135539967 2:134782397 -

(AMD) and Their Role in the Regulation of Gene Expression
Genetic Variants with Significant Association to Age-Related Macular Degeneration (AMD) and their Role in the Regulation of Gene Expression Dissertation zur Erlangung des Doktorgrades der Biomedizinischen Wissenschaften (Dr. rer. physiol.) der Fakultät für Medizin der Universität Regensburg vorgelegt von Tobias Strunz aus Marktredwitz im Jahr 2020 Genetic Variants with Significant Association to Age-Related Macular Degeneration (AMD) and their Role in the Regulation of Gene Expression Dissertation zur Erlangung des Doktorgrades der Biomedizinischen Wissenschaften (Dr. rer. physiol.) der Fakultät für Medizin der Universität Regensburg vorgelegt von Tobias Strunz aus Marktredwitz im Jahr 2020 Dekan: Prof. Dr. Dirk Hellwig Betreuer: Prof. Dr. Bernhard H.F. Weber Tag der mündlichen Prüfung: 02.12.2020 Parts of this work have already been published in peer-reviewed journals in an open access format: Strunz T, Grassmann F, Gayán J, Nahkuri S, Souza-Costa D, Maugeais C, Fauser S, Nogoceke E, Weber BHF (2018) A mega-analysis of expression quantitative trait loci (eQTL) provides insight into the regulatory architecture of gene expression variation in liver. Sci Rep 8: 5865. Strunz T, Lauwen S, Kiel C, den Hollander A, Weber BHF (2020) A transcriptome- wide association study based on 27 tissues identifies 106 genes potentially relevant for disease pathology in age-related macular degeneration. Sci Rep 10: 1584. Strunz T, Kiel C, Grassmann F, Ratnapriya R, Kwicklis M, Karlstetter M, Fauser S, Swaroop A, Arend N, Langmann T, Wolf A, Weber BHF (2020) A mega-analysis of expression quantitative trait loci in retinal tissue. PLoS Genet 16: e1008934. Kiel C, Berber P, Karlstetter M, Aslanidis A, Strunz T, Langmann T, Grassmann F, Weber BHF (2020) A Circulating MicroRNA Profile in a Laser-Induced Mouse Model of Choroidal Neovascularization. -

Genetic Landscape of Nonobstructive Azoospermia and New Perspectives for the Clinic
Journal of Clinical Medicine Review Genetic Landscape of Nonobstructive Azoospermia and New Perspectives for the Clinic Miriam Cerván-Martín 1,2, José A. Castilla 2,3,4, Rogelio J. Palomino-Morales 2,5 and F. David Carmona 1,2,* 1 Departamento de Genética e Instituto de Biotecnología, Universidad de Granada, Centro de Investigación Biomédica (CIBM), Parque Tecnológico Ciencias de la Salud, Av. del Conocimiento, s/n, 18016 Granada, Spain; [email protected] 2 Instituto de Investigación Biosanitaria ibs.GRANADA, Av. de Madrid, 15, Pabellón de Consultas Externas 2, 2ª Planta, 18012 Granada, Spain; [email protected] (J.A.C.); [email protected] (R.J.P.-M.) 3 Unidad de Reproducción, UGC Obstetricia y Ginecología, HU Virgen de las Nieves, Av. de las Fuerzas Armadas 2, 18014 Granada, Spain 4 CEIFER Biobanco—NextClinics, Calle Maestro Bretón 1, 18004 Granada, Spain 5 Departamento de Bioquímica y Biología Molecular I, Universidad de Granada, Facultad de Ciencias, Av. de Fuente Nueva s/n, 18071 Granada, Spain * Correspondence: [email protected]; Tel.: +34-958-241-000 (ext 20170) Received: 29 December 2019; Accepted: 16 January 2020; Published: 21 January 2020 Abstract: Nonobstructive azoospermia (NOA) represents the most severe expression of male infertility, involving around 1% of the male population and 10% of infertile men. This condition is characterised by the inability of the testis to produce sperm cells, and it is considered to have an important genetic component. During the last two decades, different genetic anomalies, including microdeletions of the Y chromosome, karyotype defects, and missense mutations in genes involved in the reproductive function, have been described as the primary cause of NOA in many infertile men. -

Genetics and Epigenetics in Asthma
International Journal of Molecular Sciences Review Genetics and Epigenetics in Asthma Polyxeni Ntontsi 1, Andreas Photiades 1 , Eleftherios Zervas 1 , Georgina Xanthou 2 and Konstantinos Samitas 1,2,* 1 7th Respiratory Medicine Department and Asthma Center, Athens Chest Hospital “Sotiria”, 11527 Athens, Greece; [email protected] (P.N.); [email protected] (A.P.); [email protected] (E.Z.) 2 Cellular Immunology Laboratory, Biomedical Research Foundation of the Academy of Athens, 11527 Athens, Greece; [email protected] * Correspondence: [email protected]; Tel.: +30-210-778-1720 Abstract: Asthma is one of the most common respiratory disease that affects both children and adults worldwide, with diverse phenotypes and underlying pathogenetic mechanisms poorly understood. As technology in genome sequencing progressed, scientific efforts were made to explain and pre- dict asthma’s complexity and heterogeneity, and genome-wide association studies (GWAS) quickly became the preferred study method. Several gene markers and loci associated with asthma suscep- tibility, atopic and childhood-onset asthma were identified during the last few decades. Markers near the ORMDL3/GSDMB genes were associated with childhood-onset asthma, interleukin (IL)33 and IL1RL1 SNPs were associated with atopic asthma, and the Thymic Stromal Lymphopoietin (TSLP) gene was identified as protective against the risk to TH2-asthma. The latest efforts and advances in identifying and decoding asthma susceptibility are focused on epigenetics, heritable characteristics that affect gene expression without altering DNA sequence, with DNA methylation being the most described mechanism. Other less studied epigenetic mechanisms include histone modifications and alterations of miR expression. Recent findings suggest that the DNA methylation pattern is tissue Citation: Ntontsi, P.; Photiades, A.; and cell-specific. -

The COVID-19 Gene and Drug Set Library
The COVID-19 Gene and Drug Set Library Maxim V. Kuleshov1, Daniel J.B. Clarke1, Eryk Kropiwnicki1, Kathleen M. Jagodnik1, Alon Bartal1, John E. Evangelista1, Abigail Zhou1, Laura B. Ferguson2, Alexander Lachmann1, Avi Ma’ayan1,* 1Department of Pharmacological Sciences; Mount Sinai Center for Bioinformatics; Big Data to Knowledge, Library of Integrated Network-based Cellular Signatures, Data Coordination and Integration Center (BD2K-LINCS DCIC); Knowledge Management Center for Illuminating the Druggable Genome (KMC-IDG); Icahn School of Medicine at Mount Sinai, One Gustave L. Levy Place, Box 1603, New York, NY 10029, USA 2Department of Neurology; Dell Medical School; University of Texas at Austin; 1601 Trinity Street, Bldg B, Austin, TX 78712, USA. To whom correspondence should be addressed: [email protected] Abstract The coronavirus (CoV) severe acute respiratory syndrome (SARS)-CoV-2 (COVID-19) pandemic has received rapid response by the research community to offer suggestions for repurposing of approved drugs as well as to improve our understanding of the COVID-19 viral life cycle molecular mechanisms. In a short period, tens of thousands of research preprints and other publications have emerged including those that report lists of experimentally validated drugs and compounds as potential COVID-19 therapies. In addition, gene sets from interacting COVID-19 virus-host proteins and differentially expressed genes when comparing infected to uninfected cells are being published at a fast rate. To organize this rapidly accumulating knowledge, we developed the COVID-19 Gene and Drug Set Library (https://amp.pharm.mssm.edu/covid19/), a collection of gene and drug sets related to COVID-19 research from multiple sources. -

Genetic Impacts on DNA Methylation: Research Findings and Future Perspectives
Villicaña and Bell Genome Biology (2021) 22:127 https://doi.org/10.1186/s13059-021-02347-6 REVIEW Open Access Genetic impacts on DNA methylation: research findings and future perspectives Sergio Villicaña* and Jordana T. Bell *Correspondence: [email protected] Abstract Department of Twin Research and Multiple recent studies highlight that genetic variants can have strong impacts on a Genetic Epidemiology, St. Thomas’ Hospital, King’s College London, 3rd significant proportion of the human DNA methylome. Methylation quantitative trait Floor, South Wing, Block D, London loci, or meQTLs, allow for the exploration of biological mechanisms that underlie SE1 7EH, UK complex human phenotypes, with potential insights for human disease onset and progression. In this review, we summarize recent milestones in characterizing the human genetic basis of DNA methylation variation over the last decade, including heritability findings and genome-wide identification of meQTLs. We also discuss challenges in this field and future areas of research geared to generate insights into molecular processes underlying human complex traits. Keywords: DNA methylation, Heritability, GWAS, Methylation quantitative trait loci, meQTL Introduction The complexity of the human genome lies not only in its composition of billions of base pairs, but also in the chemical modifications that make it interpretable to enzymes and other molecular factors, through epigenetic mechanisms. DNA methylation has been the most widely studied epigenetic mark since 1948 when it was first reported [1]. In humans, DNA methylation consists of the covalent addition of a methyl group to cytosine residues—predominantly at CpG sites—by a family of enzymes called DNA methyltrans- ferases (DNMTs) [2, 3]. -

Use of Deep-Learning Genomics to Discriminate Healthy Individuals from Those with Alzheimer’S Disease Or Mild Cognitive Impairment
Hindawi Behavioural Neurology Volume 2021, Article ID 3359103, 15 pages https://doi.org/10.1155/2021/3359103 Research Article Use of Deep-Learning Genomics to Discriminate Healthy Individuals from Those with Alzheimer’s Disease or Mild Cognitive Impairment Lanlan Li,1 Yeying Yang,2 Qi Zhang,1 Jiao Wang,3 Jiehui Jiang ,1 and Alzheimer’s Disease Neuroimaging Initiative4 1Institute of Biomedical Engineering, School of Communication and Information Engineering, Shanghai University, Shanghai 200444, China 2LongHua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 200032, China 3School of Life Science, Shanghai University, Shanghai 200444, China 4Indiana University School of Medicine, Indianapolis, IN 46202, USA Correspondence should be addressed to Jiehui Jiang; [email protected] Received 14 May 2021; Accepted 11 June 2021; Published 15 July 2021 Academic Editor: Muh-Shi Lin Copyright © 2021 Lanlan Li et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Objectives. Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder and the most common form of dementia in the elderly. Certain genes have been identified as important clinical risk factors for AD, and technological advances in genomic research, such as genome-wide association studies (GWAS), allow for analysis of polymorphisms and have been widely applied to studies of AD. However, shortcomings of GWAS include sensitivity to sample size and hereditary deletions, which result in low classification and predictive accuracy. Therefore, this paper proposes a novel deep-learning genomics approach and applies it to multitasking classification of AD progression, with the goal of identifying novel genetic biomarkers overlooked by traditional GWAS analysis. -

A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression
Supplementary Materials A Meta-Analysis of the Effects of High-LET Ionizing Radiations in Human Gene Expression Table S1. Statistically significant DEGs (Adj. p-value < 0.01) derived from meta-analysis for samples irradiated with high doses of HZE particles, collected 6-24 h post-IR not common with any other meta- analysis group. This meta-analysis group consists of 3 DEG lists obtained from DGEA, using a total of 11 control and 11 irradiated samples [Data Series: E-MTAB-5761 and E-MTAB-5754]. Ensembl ID Gene Symbol Gene Description Up-Regulated Genes ↑ (2425) ENSG00000000938 FGR FGR proto-oncogene, Src family tyrosine kinase ENSG00000001036 FUCA2 alpha-L-fucosidase 2 ENSG00000001084 GCLC glutamate-cysteine ligase catalytic subunit ENSG00000001631 KRIT1 KRIT1 ankyrin repeat containing ENSG00000002079 MYH16 myosin heavy chain 16 pseudogene ENSG00000002587 HS3ST1 heparan sulfate-glucosamine 3-sulfotransferase 1 ENSG00000003056 M6PR mannose-6-phosphate receptor, cation dependent ENSG00000004059 ARF5 ADP ribosylation factor 5 ENSG00000004777 ARHGAP33 Rho GTPase activating protein 33 ENSG00000004799 PDK4 pyruvate dehydrogenase kinase 4 ENSG00000004848 ARX aristaless related homeobox ENSG00000005022 SLC25A5 solute carrier family 25 member 5 ENSG00000005108 THSD7A thrombospondin type 1 domain containing 7A ENSG00000005194 CIAPIN1 cytokine induced apoptosis inhibitor 1 ENSG00000005381 MPO myeloperoxidase ENSG00000005486 RHBDD2 rhomboid domain containing 2 ENSG00000005884 ITGA3 integrin subunit alpha 3 ENSG00000006016 CRLF1 cytokine receptor like -

Identifying Sex-Speci C Genetic Effects Across 733 Traits in UK Biobank
Identifying sex-specic genetic effects across 733 traits in UK Biobank James Han Yale University Wei Jiang Department of Biostatistics, Yale School of Public Health Yixuan Ye Yale University https://orcid.org/0000-0002-2643-665X Hongyu Zhao ( [email protected] ) Yale University https://orcid.org/0000-0003-1195-9607 Article Keywords: sex-specicity, diseases, traits, polygenic risk Posted Date: July 20th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-701876/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/21 Abstract Sex-specicity has been reported in a wide range of diseases and complex traits. While sex-specic genetic effects have been documented for certain traits, the genetic mechanisms underlying sex differences in most traits remain largely unexplored. With its large sample size and wide range of diseases and traits, the UK Biobank—a large, prospective cohort study containing health history, phenotypic measurements, and genetic data for over 500,000 individuals— provides an opportunity to explore sexually dimorphic genetic architectures in a large number of traits and diseases. Here, we present a sex-specic analysis of 733 sex-stratied complex trait GWAS for 361,194 white British individuals in the UK Biobank, and report 16 traits with signicant sex-specic differences in heritability. These 16 candidate traits with sex-specic genetic effects belong to 5 distinct groups: body fat mass and distribution, blood pressure, creatinine levels, snoring, and birth weight. Using a systematic sex-specic discovery-replication analysis, we identify 47 (31 novel) loci showing sex-specic effects on the traits related with body fat mass/distribution, blood pressure, and birth weight, and discover 74 potential sex-specic biological pathways from the enrichment analyses based on associated genes from QTL analysis. -

Identification of Hub Genes and Key Pathways Associated with Atopic 3 Dermatitis Via Integrated Bioinformatics 4 Yue Sun, Angela Weihong Yang and George Binh Lenon*
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 1 July 2019 doi:10.20944/preprints201907.0018.v1 1 Article 2 Identification of Hub genes and key pathways associated with atopic 3 dermatitis via Integrated Bioinformatics 4 Yue Sun, Angela Weihong Yang and George Binh Lenon* 5 1 School of Health and Biomedical Sciences, RMIT University, Victoria, Australia 6 *Correspondence: [email protected] 7 Abstract: The exploration and identification of targets and pathways for Atopic dermatitis (AD) 8 treatment and diagnosis are critical for AD control. The conventional target exploration approach 9 such as the literature review is not satisfying in terms of efficiency and accuracy. Recently, the 10 bioinformatic approach is drawing attention for its unique advantage of high-volume data analysis 11 for target and pathway exploration; Open Targets Platform is the targets source for this study to 12 extract top 200 high-rank proteins from 3122 AD associated proteins. STRING, Cytoscape, 13 CytoHubba, ClueGo, and CluePedia function had been applied for data analysis. The KEGG 14 Mapper search & colour pathway was the pathway map resource for identified pathways; 23 key 15 hub genes (VDR, KIT, BCL2L11, NFKBIA, KRAS, IL13, JAK2, STAT3, IL21, IL4R, REL, PDGFRB, 16 FOXP3, RARA, RELB, EGFR, IL21R, MYC, CREBBP, NR3C1, IL2, JAK1, and KITLG). Additionally, 17 8 correlated pathways and the biological process had been identified; Through this study, a viable 18 approach for target and pathway exploration had been presented. The identified AD targets and 19 pathways will be tested for upcoming research for traditional Chinese medicinal herb interactions. 20 21 Keywords: Atopic Dermatitis; AD; Dermatology; target identification; pathway identification; 22 Bioinformatics; Protein-protein networks. -

Characterization of Novel Progression Factors in Castration-Resistant Prostate Cancer Based on Global Comparative Proteome Analysis
cancers Article Characterization of Novel Progression Factors in Castration-Resistant Prostate Cancer Based on Global Comparative Proteome Analysis Ann-Yae Na 1,†, Soyoung Choi 1,†, Eunju Yang 2, Kwang-Hyeon Liu 1,2,3 , Sunghwan Kim 3,4, Hyun Jin Jung 5, Youngshik Choe 5, Yun-Sok Ha 6, Tae Gyun Kwon 6,7, Jun Nyung Lee 6,* and Sangkyu Lee 1,2,3,* 1 BK21 FOUR Community-Based Intelligent Novel Drug Discovery Education Unit, College of Pharmacy, Kyungpook National University, Daegu 41566, Korea; [email protected] (A.-Y.N.); [email protected] (S.C.); [email protected] (K.-H.L.) 2 Research Institute of Pharmaceutical Sciences, Kyungpook National University, Daegu 41566, Korea; [email protected] 3 Mass Spectrometry Convergence Research Center and Green-Nano Materials Research Center, Daegu 41566, Korea; [email protected] 4 Department of Chemistry, Kyungpook National University, Daegu 41566, Korea 5 Korea Brain Research Institute, Daegu 41068, Korea; [email protected] (H.J.J.); [email protected] (Y.C.) 6 Department of Urology, School of Medicine, Kyungpook National University, Daegu 41405, Korea; [email protected] (Y.-S.H.); [email protected] (T.G.K.) 7 Joint Institute for Regenerative Medicine, Kyungpook National University, Daegu 41405, Korea * Correspondence: [email protected] (J.N.L.); [email protected] (S.L.); Tel.: +82-53-200-2675 (J.N.L.); +82-53-950-5986 (S.L.) Citation: Na, A.-Y.; Choi, S.; Yang, E.; † These authors contributed equally to this work. Liu, K.-H.; Kim, S.; Jung, H.J.; Choe, Y.; Ha, Y.-S.; Kwon, T.G.; Lee, J.N.; Simple Summary: Here, we investigated prostate cancer (PCa) tissues at each stage of progression, et al. -

Investigating the DNA Methylation Profiles of Children with Oligoarticular Juvenile Idiopathic Arthritis (JIA)
Investigating the DNA methylation profiles of children with oligoarticular juvenile idiopathic arthritis (JIA). Raul Antonio Chavez Valencia Submitted in total fulfilment of the requirements of the degree of Doctor of Philosophy Department of Paediatrics Faculty of Medicine, Dentistry and Health Sciences The University of Melbourne December, 2019 Abstract Juvenile idiopathic arthritis (JIA) is a complex autoimmune disease affecting children aged between 6 months and 16 years. JIA represents a group of 7 subtypes of disease, with the most common being oligoarticular JIA (oJIA). Despite a prevalence of up to 1 in 400, rates similar to those in T1D, JIA research is relatively sparse. Research into disease pathogenesis has largely focussed on genetic risk factors, and has also identified CD4+ T-cells as likely to mediate the autoimmune process. However, research is particularly needed regarding diagnosis and prognosis of disease and its outcomes. Currently, diagnosis is almost entirely dependent on clinical observation and history, with little in the way of biomarkers to classify patients or to guide clinical management. Epigenetics represent biological modifications to DNA and chromatin that control gene expression and chromatin structure. DNA methylation is perhaps the most accessible modification available for study, and is known to modulate immune cell function particularly amongst CD4+ T-cell subsets. A number of autoimmune diseases have reported significant DNAm associations, and have also provided intriguing data on the potential of DNAm to predict clinical outcomes. This study hypothesised that DNAm is important in oJIA pathogenesis, and potentially provides a biological basis for the diagnosis and prognosis of disease. This study utilised CD4+ T-cells and a case-control study design to analyse the associations between DNAm and oJIA, with data generated from the Illumina Infinium HumanMethylation450 BeadChip array.