Characterization of Novel Progression Factors in Castration-Resistant Prostate Cancer Based on Global Comparative Proteome Analysis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genome-Wide Screen of Cell-Cycle Regulators in Normal and Tumor Cells
bioRxiv preprint doi: https://doi.org/10.1101/060350; this version posted June 23, 2016. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Genome-wide screen of cell-cycle regulators in normal and tumor cells identifies a differential response to nucleosome depletion Maria Sokolova1, Mikko Turunen1, Oliver Mortusewicz3, Teemu Kivioja1, Patrick Herr3, Anna Vähärautio1, Mikael Björklund1, Minna Taipale2, Thomas Helleday3 and Jussi Taipale1,2,* 1Genome-Scale Biology Program, P.O. Box 63, FI-00014 University of Helsinki, Finland. 2Science for Life laboratory, Department of Biosciences and Nutrition, Karolinska Institutet, SE- 141 83 Stockholm, Sweden. 3Science for Life laboratory, Division of Translational Medicine and Chemical Biology, Department of Medical Biochemistry and Biophysics, Karolinska Institutet, S-171 21 Stockholm, Sweden To identify cell cycle regulators that enable cancer cells to replicate DNA and divide in an unrestricted manner, we performed a parallel genome-wide RNAi screen in normal and cancer cell lines. In addition to many shared regulators, we found that tumor and normal cells are differentially sensitive to loss of the histone genes transcriptional regulator CASP8AP2. In cancer cells, loss of CASP8AP2 leads to a failure to synthesize sufficient amount of histones in the S-phase of the cell cycle, resulting in slowing of individual replication forks. Despite this, DNA replication fails to arrest, and tumor cells progress in an elongated S-phase that lasts several days, finally resulting in death of most of the affected cells. -

Histone-Related Genes Are Hypermethylated in Lung Cancer
Published OnlineFirst October 1, 2019; DOI: 10.1158/0008-5472.CAN-19-1019 Cancer Genome and Epigenome Research Histone-Related Genes Are Hypermethylated in Lung Cancer and Hypermethylated HIST1H4F Could Serve as a Pan-Cancer Biomarker Shihua Dong1,Wei Li1, Lin Wang2, Jie Hu3,Yuanlin Song3, Baolong Zhang1, Xiaoguang Ren1, Shimeng Ji3, Jin Li1, Peng Xu1, Ying Liang1, Gang Chen4, Jia-Tao Lou2, and Wenqiang Yu1 Abstract Lung cancer is the leading cause of cancer-related deaths lated in all 17 tumor types from TCGA datasets (n ¼ 7,344), worldwide. Cytologic examination is the current "gold stan- which was further validated in nine different types of cancer dard" for lung cancer diagnosis, however, this has low sensi- (n ¼ 243). These results demonstrate that HIST1H4F can tivity. Here, we identified a typical methylation signature of function as a universal-cancer-only methylation (UCOM) histone genes in lung cancer by whole-genome DNA methyl- marker, which may aid in understanding general tumorigen- ation analysis, which was validated by The Cancer Genome esis and improve screening for early cancer diagnosis. Atlas (TCGA) lung cancer cohort (n ¼ 907) and was further confirmed in 265 bronchoalveolar lavage fluid samples with Significance: These findings identify a new biomarker for specificity and sensitivity of 96.7% and 87.0%, respectively. cancer detection and show that hypermethylation of histone- More importantly, HIST1H4F was universally hypermethy- related genes seems to persist across cancers. Introduction to its low specificity, LDCT is far from satisfactory as a screening tool for clinical application, similar to other currently used cancer Lung cancer is one of the most common malignant tumors and biomarkers, such as carcinoembryonic antigen (CEA), neuron- the leading cause of cancer-related deaths worldwide (1, 2). -
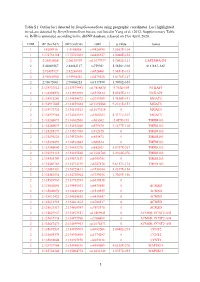
1 Table S1. Outlier Loci Detected by Deepgenomescan Using
Table S1. Outlier loci detected by DeepGenomeScan using geographic coordinates. Loci highlighted in red are detected by DeepGenomeScan but are not listed in Yang et al. (2012; Supplementary Table 4). RsID is annotated according to the dbSNP database released on 21st April, 2020. CHR BP (Grch37) BP (Grch38) rsID p.value Genes 1 1:4208918 1:4148858 rs9426495 3.8582E-104 1 1:175738168 1:175769032 rs6425357 2.0088E-105 2 2:20310668 2:20110907 rs11679737 1.2002E-111 LAPTM4A-DT 2 2:40289267 2:40062127 rs759361 5.5658E-104 SLC8A1-AS1 2 2:82495127 2:82268003 rs6726401 3.9451E-103 2 2:96660300 2:95994552 rs2579520 1.8176E-127 2 2:96672001 2:96006253 rs1917890 3.7698E-104 2 2:134333012 2:133575441 rs17816830 9.702E-105 NCKAP5 2 2:134350570 2:133592999 rs6715224 5.8745E-111 NCKAP5 2 2:134912243 2:134154672 rs2139309 1.7418E-191 MGAT5 2 2:134917005 2:134159434 rs11692586 9.2121E-151 MGAT5 2 2:134972732 2:134215161 rs11679218 0 MGAT5 2 2:134979966 2:134222395 rs1965183 5.3171E-107 MGAT5 2 2:135260071 2:134502500 rs503562 2.057E-153 TMEM163 2 2:135280039 2:134522468 rs579670 3.1477E-118 TMEM163 2 2:135285279 2:134527708 rs512375 0 TMEM163 2 2:135290221 2:134532650 rs655472 0 TMEM163 2 2:135290453 2:134532882 rs666614 0 TMEM163 2 2:135340840 2:134583270 rs842361 5.6717E-237 TMEM163 2 2:135393110 2:134635540 rs11684785 2.2164E-276 TMEM163 2 2:135430709 2:134673139 rs6745983 0 TMEM163 2 2:135469769 2:134712199 rs6747870 9.6117E-114 TMEM163 2 2:135483381 2:134725811 rs3739034 5.0357E-154 2 2:135483534 2:134725964 rs3739036 3.7269E-156 2 2:135539967 2:134782397 -

Download These Multimorbidities (
Dong et al. Genome Medicine (2021) 13:110 https://doi.org/10.1186/s13073-021-00927-6 RESEARCH Open Access A global overview of genetically interpretable multimorbidities among common diseases in the UK Biobank Guiying Dong1,2, Jianfeng Feng1,2,3, Fengzhu Sun4, Jingqi Chen1,2,3* and Xing-Ming Zhao1,2,3* Abstract Background: Multimorbidities greatly increase the global health burdens, but the landscapes of their genetic risks have not been systematically investigated. Methods: We used the hospital inpatient data of 385,335 patients in the UK Biobank to investigate the multimorbid relations among 439 common diseases. Post-GWAS analyses were performed to identify multimorbidity shared genetic risks at the genomic loci, network, as well as overall genetic architecture levels. We conducted network decomposition for the networks of genetically interpretable multimorbidities to detect the hub diseases and the involved molecules and functions in each module. Results: In total, 11,285 multimorbidities among 439 common diseases were identified, and 46% of them were genetically interpretable at the loci, network, or overall genetic architecture levels. Multimorbidities affecting the same and different physiological systems displayed different patterns of the shared genetic components, with the former more likely to share loci-level genetic components while the latter more likely to share network-level genetic components. Moreover, both the loci- and network-level genetic components shared by multimorbidities converged on cell immunity, protein metabolism, and gene silencing. Furthermore, we found that the genetically interpretable multimorbidities tend to form network modules, mediated by hub diseases and featuring physiological categories. Finally, we showcased how hub diseases mediating the multimorbidity modules could help provide useful insights for the genetic contributors of multimorbidities. -

The HIST1 Locus Escapes Reprogramming in Cloned Bovine Embryos
INVESTIGATION The HIST1 Locus Escapes Reprogramming in Cloned Bovine Embryos Byungkuk Min,*,† Sunwha Cho,* Jung Sun Park,* Kyuheum Jeon,*,† and Yong-Kook Kang*,†,1 *Development and Differentiation Research Center, Korea Research Institute of Bioscience and Biotechnology (KRIBB), Daejeon, 305-806, South Korea, †Department of Functional Genomics, University of Science and Technology, Daejeon, 305-350, South Korea ORCID ID: 0000-0002-5419-6964 (B.M.) ABSTRACT Epigenetic reprogramming is necessary in somatic cell nuclear transfer (SCNT) embryos in KEYWORDS order to erase the differentiation-associated epigenetic marks of donor cells. However, such epigenetic RNA-seq memories often persist throughout the course of clonal development, thus decreasing cloning efficiency. SCNT Here, we explored reprogramming-refractory regions in bovine SCNT blastocyst transcriptomes. We HIST1 cluster observed that histone genes residing in the 1.5 Mb spanning the cow HIST1 cluster were coordinately reprogramming downregulated in SCNT blastocysts. In contrast, both the nonhistone genes of this cluster, and histone epigenetics genes elsewhere remained unaffected. This indicated that the downregulation was specifictoHIST1 histone HDAC inhibitor genes. We found that, after trichostatin A treatment, HIST1 histone genes were derepressed, and DNA DNA methylation methylation at their promoters was decreased to the level of in vitro fertilization embryos. Therefore, our results indicate that the reduced expression of HIST1 histone genes is a consequence of poor epigenetic reprogramming in SCNT blastocysts. Successful cloning by somatic cell nuclear transfer (SCNT) depends and SCNT blastocysts (Min et al. 2015). Additionally, we also reported onaccuratereprogrammingprocessesinwhichthedifferentiatedstateof features unique to SCNT blastocysts, including consistently aberrant the donor cell genome drifts to a totipotent, embryonic ground state expression of genes that function in either trophectoderm development (Kang et al. -

A Multiprotein Occupancy Map of the Mrnp on the 3 End of Histone
Downloaded from rnajournal.cshlp.org on October 6, 2021 - Published by Cold Spring Harbor Laboratory Press A multiprotein occupancy map of the mRNP on the 3′ end of histone mRNAs LIONEL BROOKS III,1 SHAWN M. LYONS,2 J. MATTHEW MAHONEY,1 JOSHUA D. WELCH,3 ZHONGLE LIU,1 WILLIAM F. MARZLUFF,2 and MICHAEL L. WHITFIELD1 1Department of Genetics, Dartmouth Geisel School of Medicine, Hanover, New Hampshire 03755, USA 2Integrative Program for Biological and Genome Sciences, University of North Carolina, Chapel Hill, North Carolina 27599, USA 3Department of Computer Science, University of North Carolina, Chapel Hill, North Carolina 27599, USA ABSTRACT The animal replication-dependent (RD) histone mRNAs are coordinately regulated with chromosome replication. The RD-histone mRNAs are the only known cellular mRNAs that are not polyadenylated. Instead, the mature transcripts end in a conserved stem– loop (SL) structure. This SL structure interacts with the stem–loop binding protein (SLBP), which is involved in all aspects of RD- histone mRNA metabolism. We used several genomic methods, including high-throughput sequencing of cross-linked immunoprecipitate (HITS-CLIP) to analyze the RNA-binding landscape of SLBP. SLBP was not bound to any RNAs other than histone mRNAs. We performed bioinformatic analyses of the HITS-CLIP data that included (i) clustering genes by sequencing read coverage using CVCA, (ii) mapping the bound RNA fragment termini, and (iii) mapping cross-linking induced mutation sites (CIMS) using CLIP-PyL software. These analyses allowed us to identify specific sites of molecular contact between SLBP and its RD-histone mRNA ligands. We performed in vitro crosslinking assays to refine the CIMS mapping and found that uracils one and three in the loop of the histone mRNA SL preferentially crosslink to SLBP, whereas uracil two in the loop preferentially crosslinks to a separate component, likely the 3′hExo. -

Research Article Common Expression Quantitative Trait Loci Shared by Histone Genes
Hindawi International Journal of Genomics Volume 2017, Article ID 6202567, 14 pages https://doi.org/10.1155/2017/6202567 Research Article Common Expression Quantitative Trait Loci Shared by Histone Genes Hanseol Kim, Yujin Suh, and Chaeyoung Lee Department of Bioinformatics and Life Science, Soongsil University, Seoul, Republic of Korea Correspondence should be addressed to Chaeyoung Lee; [email protected] Received 29 May 2017; Revised 26 July 2017; Accepted 2 August 2017; Published 27 August 2017 Academic Editor: Marco Gerdol Copyright © 2017 Hanseol Kim et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. A genome-wide association study (GWAS) was conducted to examine expression quantitative trait loci (eQTLs) for histone genes. We examined common eQTLs for multiple histone genes in 373 European lymphoblastoid cell lines (LCLs). A linear regression model was employed to identify single-nucleotide polymorphisms (SNPs) associated with expression of the histone genes, and the number of eQTLs was determined by linkage disequilibrium analysis. Additional associations of the identified eQTLs with other genes were also examined. We identified 31 eQTLs for 29 histone genes through genome-wide analysis using 29 histone genes (P <297 × 10−10). Among them, 12 eQTLs were associated with the expression of multiple histone genes. Transcriptome- wide association analysis using the identified eQTLs showed their associations with additional 80 genes (P <475 × 10−6). In particular, expression of RPPH1, SCARNA2, and SCARNA7 genes was associated with 26, 25, and 23 eQTLs, respectively. -

(AMD) and Their Role in the Regulation of Gene Expression
Genetic Variants with Significant Association to Age-Related Macular Degeneration (AMD) and their Role in the Regulation of Gene Expression Dissertation zur Erlangung des Doktorgrades der Biomedizinischen Wissenschaften (Dr. rer. physiol.) der Fakultät für Medizin der Universität Regensburg vorgelegt von Tobias Strunz aus Marktredwitz im Jahr 2020 Genetic Variants with Significant Association to Age-Related Macular Degeneration (AMD) and their Role in the Regulation of Gene Expression Dissertation zur Erlangung des Doktorgrades der Biomedizinischen Wissenschaften (Dr. rer. physiol.) der Fakultät für Medizin der Universität Regensburg vorgelegt von Tobias Strunz aus Marktredwitz im Jahr 2020 Dekan: Prof. Dr. Dirk Hellwig Betreuer: Prof. Dr. Bernhard H.F. Weber Tag der mündlichen Prüfung: 02.12.2020 Parts of this work have already been published in peer-reviewed journals in an open access format: Strunz T, Grassmann F, Gayán J, Nahkuri S, Souza-Costa D, Maugeais C, Fauser S, Nogoceke E, Weber BHF (2018) A mega-analysis of expression quantitative trait loci (eQTL) provides insight into the regulatory architecture of gene expression variation in liver. Sci Rep 8: 5865. Strunz T, Lauwen S, Kiel C, den Hollander A, Weber BHF (2020) A transcriptome- wide association study based on 27 tissues identifies 106 genes potentially relevant for disease pathology in age-related macular degeneration. Sci Rep 10: 1584. Strunz T, Kiel C, Grassmann F, Ratnapriya R, Kwicklis M, Karlstetter M, Fauser S, Swaroop A, Arend N, Langmann T, Wolf A, Weber BHF (2020) A mega-analysis of expression quantitative trait loci in retinal tissue. PLoS Genet 16: e1008934. Kiel C, Berber P, Karlstetter M, Aslanidis A, Strunz T, Langmann T, Grassmann F, Weber BHF (2020) A Circulating MicroRNA Profile in a Laser-Induced Mouse Model of Choroidal Neovascularization. -

Supplemental Data.Pdf
Supplementary material -Table of content Supplementary Figures (Fig 1- Fig 6) Supplementary Tables (1-13) Lists of genes belonging to distinct biological processes identified by GREAT analyses to be significantly enriched with UBTF1/2-bound genes Supplementary Table 14 List of the common UBTF1/2 bound genes within +/- 2kb of their TSSs in NIH3T3 and HMECs. Supplementary Table 15 List of gene identified by microarray expression analysis to be differentially regulated following UBTF1/2 knockdown by siRNA Supplementary Table 16 List of UBTF1/2 binding regions overlapping with histone genes in NIH3T3 cells Supplementary Table 17 List of UBTF1/2 binding regions overlapping with histone genes in HMEC Supplementary Table 18 Sequences of short interfering RNA oligonucleotides Supplementary Table 19 qPCR primer sequences for qChIP experiments Supplementary Table 20 qPCR primer sequences for reverse transcription-qPCR Supplementary Table 21 Sequences of primers used in CHART-PCR Supplementary Methods Supplementary Fig 1. (A) ChIP-seq analysis of UBTF1/2 and Pol I (POLR1A) binding across mouse rDNA. UBTF1/2 is enriched at the enhancer and promoter regions and along the entire transcribed portions of rDNA with little if any enrichment in the intergenic spacer (IGS), which separates the rDNA repeats. This enrichment coincides with the distribution of the largest subunit of Pol I (POLR1A) across the rDNA. All sequencing reads were mapped to the published complete sequence of the mouse rDNA repeat (Gene bank accession number: BK000964). The graph represents the frequency of ribosomal sequences enriched in UBTF1/2 and Pol I-ChIPed DNA expressed as fold change over those of input genomic DNA. -

Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name histone cluster 1, H2ak Gene Symbol HIST1H2AK Organism Human Gene Summary Histones are basic nuclear proteins that are responsible for the nucleosome structure of the chromosomal fiber in eukaryotes. Two molecules of each of the four core histones (H2A H2B H3 and H4) form an octamer around which approximately 146 bp of DNA is wrapped in repeating units called nucleosomes. The linker histone H1 interacts with linker DNA between nucleosomes and functions in the compaction of chromatin into higher order structures. This gene is intronless and encodes a member of the histone H2A family. Transcripts from this gene lack polyA tails but instead contain a palindromic termination element. This gene is found in the small histone gene cluster on chromosome 6p22-p21.3. Gene Aliases H2A/d, H2AFD, HIST1H2AG, HIST1H2AJ, HIST1H2AL, HIST1H2AM RefSeq Accession No. NC_000006.11, NG_000009.2, NT_007592.15 UniGene ID Hs.734717 Ensembl Gene ID ENSG00000184348 Entrez Gene ID 8330 Assay Information Unique Assay ID qHsaCEP0054922 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000330180 Amplicon Context Sequence TTGGTTTGGACCGAGGTATGAGTAATGAACTGCTCCAGCCCCGCTACTTGCCCT TGGCCTTGTGGTGGCTCTCAGTTTTCTTAGGCAGCAGCACGGCCTGGA Amplicon Length (bp) 72 Chromosome Location 6:27805682-27805783 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9996 cDNA Cq 21.18 Page 1/5 PrimePCR™Assay Validation Report cDNA -

Genetic Landscape of Nonobstructive Azoospermia and New Perspectives for the Clinic
Journal of Clinical Medicine Review Genetic Landscape of Nonobstructive Azoospermia and New Perspectives for the Clinic Miriam Cerván-Martín 1,2, José A. Castilla 2,3,4, Rogelio J. Palomino-Morales 2,5 and F. David Carmona 1,2,* 1 Departamento de Genética e Instituto de Biotecnología, Universidad de Granada, Centro de Investigación Biomédica (CIBM), Parque Tecnológico Ciencias de la Salud, Av. del Conocimiento, s/n, 18016 Granada, Spain; [email protected] 2 Instituto de Investigación Biosanitaria ibs.GRANADA, Av. de Madrid, 15, Pabellón de Consultas Externas 2, 2ª Planta, 18012 Granada, Spain; [email protected] (J.A.C.); [email protected] (R.J.P.-M.) 3 Unidad de Reproducción, UGC Obstetricia y Ginecología, HU Virgen de las Nieves, Av. de las Fuerzas Armadas 2, 18014 Granada, Spain 4 CEIFER Biobanco—NextClinics, Calle Maestro Bretón 1, 18004 Granada, Spain 5 Departamento de Bioquímica y Biología Molecular I, Universidad de Granada, Facultad de Ciencias, Av. de Fuente Nueva s/n, 18071 Granada, Spain * Correspondence: [email protected]; Tel.: +34-958-241-000 (ext 20170) Received: 29 December 2019; Accepted: 16 January 2020; Published: 21 January 2020 Abstract: Nonobstructive azoospermia (NOA) represents the most severe expression of male infertility, involving around 1% of the male population and 10% of infertile men. This condition is characterised by the inability of the testis to produce sperm cells, and it is considered to have an important genetic component. During the last two decades, different genetic anomalies, including microdeletions of the Y chromosome, karyotype defects, and missense mutations in genes involved in the reproductive function, have been described as the primary cause of NOA in many infertile men. -

Ontology-Driven Pathway Data Integration
©Copyright 2019 Lucy Lu Wang Ontology-driven pathway data integration Lucy Lu Wang A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Washington 2019 Reading Committee: John H. Gennari, Chair Neil F. Abernethy Paul K. Crane Program Authorized to Offer Degree: Biomedical & Health Informatics University of Washington Abstract Ontology-driven pathway data integration Lucy Lu Wang Chair of the Supervisory Committee: Graduate Program Director & Associate Professor John H. Gennari Biomedical Informatics and Medical Education Biological pathways are useful tools for understanding human physiology and disease pathogenesis. Pathway analysis can be used to detect genes and functions associated with complex disease pheno- types. When performing pathway analysis, researchers take advantage of multiple pathway datasets, combining pathways from different pathway databases. Pathways from different databases do not eas- ily inter-operate, and the resulting combined pathway dataset can suffer from redundancy or reduced interpretability. Ontologies have been used to organize pathway data and eliminate redundancy. I generated clus- ters of semantically similar pathways by mapping pathways from seven databases to classes of one such ontology, the Pathway Ontology (PW). I then produced a typology of differences between pathways by summarizing the differences in content and knowledge representation between databases. Using the typology, I optimized an entity and graph-based network alignment algorithm for aligning pathways between databases. The algorithm was applied to clusters of semantically similar pathways to generate normalized pathways for each PW class. These normalized pathways were used to produce normal- ized gene sets for gene set enrichment analysis (GSEA). I evaluated these normalized gene sets against baseline gene sets in GSEA using four public gene expression datasets.