AAAATTGA: a Uniquely Conserved Sequence Motif in the Promoter
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Representation for Discovery of Protein Motifs
From: ISMB-93 Proceedings. Copyright © 1993, AAAI (www.aaai.org). All rights reserved. Representation for Discovery of Protein Motifs Darrell ConklinT, Suzanne Fortier~, Janice Glasgowt Departments~t of Computing and Information Sciencet and Chemistry Queen’s University Kingston, Ontario, Canada K7L 3N6 [email protected] Abstract pattern of amino acid residues. Protein motifs can be roughly classified into four categories. Sequence There are several dimensions and levels of com- motifs are linear strings of residues with an implicit plexity in which information on protein mo- topological ordering. Sequence-structure motifs are tifs may be available. For example, one- sequence motifs with secondary structure identifiers at- dimensional sequence motifs may be associated tached to one or more residues in the motif. Structure with secondary structure identifiers. Alterna- motifs are 3D structural objects, described by posi- tively, three-dimensional information on polypep- tions of residue objects in 3D Euclidian space. Apart tide segments may be used to induce prototypical from a topological linear ordering on the residues, three-dimensional structure templates. This pa- structure motifs are free of sequence information. Fi- per surveys various representations encountered nally, structure-sequence motifs are combined 1D- in the protein motif discovery literature. Many 3D structures that associate sequence information with of the representations are based on incompatible a structure motif. Figure 1 illustrates these four types semantics, making difficult the comparison and of protein motifs, along with somefurther subclassifi- combination of previous results. To make better cations which are elaborated upon later in the paper. use of machine learning techniques and to pro- The first three motif types are discussed by Thornton vide for an integrated knowledge representation and Gardner (1989); the structure-sequence motif will framework, a general representation language -- be presented in this paper. -

Preinitiation Complex
Science Highlight – June 2011 Transcription Starts Here: Structural Models of a “Minimal” Preinitiation Complex RNA polymerase II (pol II) plays a central role in the regulation of gene expression. Pol II is the enzyme responsible for synthesizing all the messenger RNA (mRNA) and most of the small nuclear RNA (snRNA) in eukaryotes. One of the key questions for transcription is how pol II decides where to start on the genomic DNA to specifically and precisely turn on a gene. This is achieved during transcription initiation by concerted actions of the core enzyme pol II and a myriad of transcription factors including five general transcription factors, known as TFIIB, -D, -E, -F, -H, which together form a giant transcription preinitiation complex on a promoter prior to transcription. One of the most prominent core promoter DNA elements is the TATA box, usually directing transcription of tissue-specific genes. TATA-box binding protein (TBP), a key component of TFIID, recognizes the TATA DNA sequence. Based on the previous crystallographic studies, the TATA box DNA is bent by nearly 90 degree through the binding of TBP (1). This striking structural feature is thought to serve as a physical landmark for the location of active genes on the genome. In addition, the location of the TATA box at least in part determines the transcription start site (TSS) in most eukaryotes, including humans. The distance between the TATA box and the TSS is conserved at around 30 base pairs. TBP does not contact pol II directly and the TATA-containing promoter must be directed to the core enzyme through another essential transcription factor TFIIB. -

Molecular Biology and Applied Genetics
MOLECULAR BIOLOGY AND APPLIED GENETICS FOR Medical Laboratory Technology Students Upgraded Lecture Note Series Mohammed Awole Adem Jimma University MOLECULAR BIOLOGY AND APPLIED GENETICS For Medical Laboratory Technician Students Lecture Note Series Mohammed Awole Adem Upgraded - 2006 In collaboration with The Carter Center (EPHTI) and The Federal Democratic Republic of Ethiopia Ministry of Education and Ministry of Health Jimma University PREFACE The problem faced today in the learning and teaching of Applied Genetics and Molecular Biology for laboratory technologists in universities, colleges andhealth institutions primarily from the unavailability of textbooks that focus on the needs of Ethiopian students. This lecture note has been prepared with the primary aim of alleviating the problems encountered in the teaching of Medical Applied Genetics and Molecular Biology course and in minimizing discrepancies prevailing among the different teaching and training health institutions. It can also be used in teaching any introductory course on medical Applied Genetics and Molecular Biology and as a reference material. This lecture note is specifically designed for medical laboratory technologists, and includes only those areas of molecular cell biology and Applied Genetics relevant to degree-level understanding of modern laboratory technology. Since genetics is prerequisite course to molecular biology, the lecture note starts with Genetics i followed by Molecular Biology. It provides students with molecular background to enable them to understand and critically analyze recent advances in laboratory sciences. Finally, it contains a glossary, which summarizes important terminologies used in the text. Each chapter begins by specific learning objectives and at the end of each chapter review questions are also included. -

Introduction to Sequence Motif Discovery and Search the Complete
Introduction to sequence motif discovery and search EMBNET course Bioinformatics of transcriptional regulation Jan 28 2008 Christoph Schmid The complete genome era Components of transcriptional regulation Distal transcription-factor binding sites (enhancer) cis-regulatory modules Wasserman 5, 276-287 (2004) DNA-protein interaction Allen et al. © 1998 EMBL Jordan et al. © 1991 Prentice-Hall Sequence motifs sequence variants of site: ..A T C G C A.. ..T T G G A C.. ..T T G G T G.. ..A T C G G T.. matrix: A 2 0 0 0 1 1 (simplest version) C 0 0 2 0 1 1 G 0 0 2 4 1 1 T 2 4 0 0 1 1 + cutoff ! sequence logo: Title Representation of the binding specificity by a scoring matrix (also referred to as weight matrix) 1 2 3 4 5 6 7 8 9 A -10 -10 -14 -12 -10 5 -2 -10 -6 C 5 -10 -13 -13 -7 -15 -13 3 -4 G -3 -14 -13 -11 5 -12 -13 2 -7 T -5 5 5 5 -10 -9 5 -11 5 Strong C T T T G A T C T Binding site 5 + 5 + 5 + 5 + 5 + 5 + 5 + 3 + 5 = 43 Random A C G T A C G T A Sequence -10 -10 -13 + 5 -10 -15 -13 -11 - 6 = -83 Biophysical interpretation of protein binding sites Columns of a weight matrix characterize the specificity of base-pair acceptor sites on the protein surface. Weight matrix elements represent negated energy contributions to the total binding energy → weight matrix score inversely proportional to binding energy Motif search: Statistical over-representation of genomic sequence motifs Conserved motifs represent protein binding sites Putative conservation in nucleotide sequence (motif) position relative to: TSS other binding sites (protein complexes) -

Tnlo-Encoded Tet Repressor Can Regulate an Operator-Containing
Proc. Nati. Acad. Sci. USA Vol. 85, pp. 1394-1397, March 1988 Biochemistry TnlO-encoded tet repressor can regulate an operator-containing plant promoter (cauliflower mosaic virus 35S promoter/electroporation/transient chloramphenicol acetyltransferase assays) CHRISTIANE GATZ* AND PETER H. QUAILt Departments of Botany and Genetics, University of Wisconsin, Madison, WI 53706 Communicated by Folke Skoog, October 26, 1987 (receivedfor review July S, 1987) ABSTRACT The TnlO-encoded tet repressor-operator The TnlO-encoded tet repressor regulates the expression system was used to regulate transcription from the cauliflower of the Tc resistance operon by binding to nearly identical mosaic virus (CaMV) 35S promoter. Expression was moni- operator sequences that overlap with three divergent pro- tored in a transient assay system by using electric field- moters (14, 15). The genes of the tet operon are only mediated gene transfer ("electroporation") into tobacco pro- transcribed in the presence of the inducer Tc, which pre- toplasts. The tet repressor, being expressed in the plant cells vents the repressor from binding to its operator sequences. under the control of eukaryotic transcription signals, blocks The tet repressor was chosen for regulating a plant promoter transcription of a CaMV 35S promoter chloramphenicol ace- for two reasons. (i) With a native molecular mass of 48 kDa, tyltransferase (cat) fusion gene when the two tet operators diffusion into the nucleus seemed likely (16). (ii) The high flank the "TATA" box. In the presence of the inducer equilibrium association constant of the repressor-inducer tetracycline, expression is restored to full activity. Location of complex ensures efficient induction at sublethal Tc concen- the operators 21 base pairs downstream of the transcription trations (17), thus making the system useful as an on/off start site does not significantly affect transcription in the switch for the specific regulation of transferred genes. -

Tools for Motif and Pattern Searching
TOOLS FOR MOTIF AND PATTERN SEARCHING Prat Thiru OUTLINE • What are motifs? • Algorithms Used and Programs Available • Workflow and Strategies • MEME/MAST Demo (online and command line) Protein Motifs DNA Motifs MEME Output Definitions • Motif: Conserved regions of protein or DNA sequences • Pattern: Qualitative description of a motif eg. regular expression C[AT]AAT[CG]X •Profile: Quantitative description of a motif eg. position weight matrix Patterns • Regular Expression Symbols ¾[ ] – OR eg. [GA] means G or A ¾{ } – NOT eg. {P,V} means not P or V ¾( ) – repeats eg. A(3) means AAA ¾X or N or “.” – any • Complex patterns representation difficult • Loose frequency information eg. [AT] vs 20%A 80%T Profiles Sequence Logos Algorithms • Enumeration • Probabilistic Optimization • Deterministic Optimization 1. Identify motifs 2. Build a consensus Enumeration • Exhaustive search: word counting method, count all n-mers and look for overrepresentation • Less likely to get stuck in a local optimum • Computationally expensive ¾YMF http://wingless.cs.washington.edu/YMF/YMFWeb/YMFInput.pl ¾Weeder http://159.149.109.9/weederaddons/locator.html Probabilistic Optimization • Uses a Gibbs sampling approach • One n-mer from each sequence is randomly picked to determine initial model. In subsequent iterations, one sequence, i, is removed and the model is recalculated. Pick a new location of motif in sequence i iterate until convergence • Assumes most sequences will have the motif ¾ AlignAce http://atlas.med.harvard.edu/cgi-bin/alignace.pl ¾ Gibbs Motif Sampler http://bayesweb.wadsworth.org/gibbs/gibbs.html Deterministic Optimization • Based on expectation maximization (EM) • EM: iteratively estimates the likelihood given the data that is present I. -

Degsampler: Gibbs Sampling Strategy for Predicting E3-Binding Sites with Position-Specific Prior Information
DegSampler: Gibbs sampling strategy for predicting E3-binding sites with position-specic prior information Osamu Maruyama ( [email protected] ) Kyushu University https://orcid.org/0000-0002-5760-1507 Fumiko Matsuzaki Kyushu University Research article Keywords: motif, substrate protein, E3 ubiquitin ligase, prior, posterior, disorder, collapsed, Gibbs sampling, DegSampler Posted Date: November 6th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-101967/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Maruyama and Matsuzaki RESEARCH DegSampler: Gibbs sampling strategy for predicting E3-binding sites with position-specific prior information Osamu Maruyama1* and Fumiko Matsuzaki2 Background Eukaryotic cells have two major pathways for degrad- ing proteins in order to control cellular processes and maintain intracellular homeostasis. One of the path- ways is autophagy, a catabolic process that deliv- ers intracellular components to lysosomes or vacuoles Abstract [1]. The other pathway is the ubiquitin-proteasome Background: The ubiquitin-proteasome system is system, which degrades polyubiquitin-tagged pro- a pathway in eukaryotic cells for degrading teins through the proteasomal machinery [2]. In the polyubiquitin-tagged proteins through the ubiquitin-proteasome system, an E3 ubiquitin ligase proteasomal machinery to control various cellular (hereinafter E3) selectively recognizes and binds to processes and maintain intracellular homeostasis. specific regions of the target substrate proteins. These In this system, the E3 ubiquitin ligase (hereinafter binding sites are called degrons [3]. E3) plays an important role in selectively It is important to identify the E3s and substrate recognizing and binding to specific regions of its proteins that interact with one another to character- substrate proteins. -

Bioinformatics: a Practical Guide to the Analysis of Genes and Proteins, Second Edition Andreas D
BIOINFORMATICS A Practical Guide to the Analysis of Genes and Proteins SECOND EDITION Andreas D. Baxevanis Genome Technology Branch National Human Genome Research Institute National Institutes of Health Bethesda, Maryland USA B. F. Francis Ouellette Centre for Molecular Medicine and Therapeutics Children’s and Women’s Health Centre of British Columbia University of British Columbia Vancouver, British Columbia Canada A JOHN WILEY & SONS, INC., PUBLICATION New York • Chichester • Weinheim • Brisbane • Singapore • Toronto BIOINFORMATICS SECOND EDITION METHODS OF BIOCHEMICAL ANALYSIS Volume 43 BIOINFORMATICS A Practical Guide to the Analysis of Genes and Proteins SECOND EDITION Andreas D. Baxevanis Genome Technology Branch National Human Genome Research Institute National Institutes of Health Bethesda, Maryland USA B. F. Francis Ouellette Centre for Molecular Medicine and Therapeutics Children’s and Women’s Health Centre of British Columbia University of British Columbia Vancouver, British Columbia Canada A JOHN WILEY & SONS, INC., PUBLICATION New York • Chichester • Weinheim • Brisbane • Singapore • Toronto Designations used by companies to distinguish their products are often claimed as trademarks. In all instances where John Wiley & Sons, Inc., is aware of a claim, the product names appear in initial capital or ALL CAPITAL LETTERS. Readers, however, should contact the appropriate companies for more complete information regarding trademarks and registration. Copyright ᭧ 2001 by John Wiley & Sons, Inc. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic or mechanical, including uploading, downloading, printing, decompiling, recording or otherwise, except as permitted under Sections 107 or 108 of the 1976 United States Copyright Act, without the prior written permission of the Publisher. -
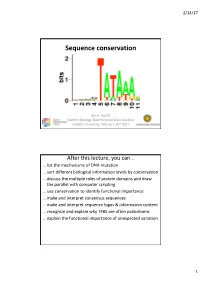
Sequence Conservation
2/14/17 Sequence conservation Bas E. Dutilh Systems Biology: Bioinformatic Data Analysis Utrecht University, FeBruary 16th 2017 After this lecture, you can… … list the mechanisms of DNA mutation … sort different biological information levels By conservation … discuss the multiple roles of protein domains and draw the parallel with computer scripting … use conservation to identify functional importance … make and interpret consensus sequences … make and interpret sequence logos & information content … recognize and explain why TFBS are often palindromic … explain the functional importance of unexpected variation 1 2/14/17 Evolution • Replication (copy) • Mutation (modify) • Selection (fitness) Time Mutations • Nucleotide suBstitutions – Replication error – Physical or chemical reaction • Insertions or deletions (indels) – Unequal crossing over during meiosis – Replication slippage • Inversions or rearrangements • Duplications of: – Partial or whole gene – Partial (polysomy) or whole chromosome (aneuploidy, polysomy) – Whole genome (polyploidy) • Horizontal gene transfer (HGT) – Conjugation (direct transfer between Bacteria) – Transformation by naturally competent Bacteria – Transduction by bacteriophages 2 2/14/17 Phenotypic/genotypic similarity • We exploit similarity to… …identify homology (shared ancestry) …determine evolutionary relationships …transfer functional information • Sequences (genotype) rarely converge • Functions (phenotype) can converge • Analogy • Homology – Similar function – Similar ancestry Low-complexity regions • -

Alignment, Clustering and Extraction of Structured Motifs in DNA Promoter Sequences
Syracuse University SURFACE Electrical Engineering and Computer Science - Dissertations College of Engineering and Computer Science 8-2012 Alignment, Clustering and Extraction of Structured Motifs in DNA Promoter Sequences Faisal Abdulmalek Alobaid Syracuse University Follow this and additional works at: https://surface.syr.edu/eecs_etd Part of the Computer Engineering Commons Recommended Citation Alobaid, Faisal Abdulmalek, "Alignment, Clustering and Extraction of Structured Motifs in DNA Promoter Sequences" (2012). Electrical Engineering and Computer Science - Dissertations. 322. https://surface.syr.edu/eecs_etd/322 This Dissertation is brought to you for free and open access by the College of Engineering and Computer Science at SURFACE. It has been accepted for inclusion in Electrical Engineering and Computer Science - Dissertations by an authorized administrator of SURFACE. For more information, please contact [email protected]. ABSTRACT A simple motif is a short DNA sequence found in the promoter region and believed to act as a binding site for a transcription factor protein. A structured motif is a sequence of simple motifs (boxes) separated by short sequences (gaps). Biologists theorize that the presence of these motifs play a key role in gene expression regulation. Discovering these patterns is an important step towards understanding protein-gene and gene-gene interaction thus facilitates the building of accurate gene regulatory network models. DNA sequence motif extraction is an important problem in bioinformatics. Many studies have proposed algorithms to solve the problem instance of simple motif extraction. Only in the past decade has the more complex structured motif extraction problem been examined by researchers. The problem is inherently challenging as structured motif patterns are segmented into several boxes separated by variable size gaps for each instance. -

ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond
International Journal of Molecular Sciences Review ER-Resident Transcription Factor Nrf1 Regulates Proteasome Expression and Beyond Jun Hamazaki * and Shigeo Murata Laboratory of Protein Metabolism, Graduate School of Pharmaceutical Sciences, the University of Tokyo, Tokyo 1130033, Japan; [email protected] * Correspondence: [email protected] Received: 4 May 2020; Accepted: 20 May 2020; Published: 23 May 2020 Abstract: Protein folding is a substantively error prone process, especially when it occurs in the endoplasmic reticulum (ER). The highly exquisite machinery in the ER controls secretory protein folding, recognizes aberrant folding states, and retrotranslocates permanently misfolded proteins from the ER back to the cytosol; these misfolded proteins are then degraded by the ubiquitin–proteasome system termed as the ER-associated degradation (ERAD). The 26S proteasome is a multisubunit protease complex that recognizes and degrades ubiquitinated proteins in an ATP-dependent manner. The complex structure of the 26S proteasome requires exquisite regulation at the transcription, translation, and molecular assembly levels. Nuclear factor erythroid-derived 2-related factor 1 (Nrf1; NFE2L1), an ER-resident transcription factor, has recently been shown to be responsible for the coordinated expression of all the proteasome subunit genes upon proteasome impairment in mammalian cells. In this review, we summarize the current knowledge regarding the transcriptional regulation of the proteasome, as well as recent findings concerning the regulation of Nrf1 transcription activity in ER homeostasis and metabolic processes. Keywords: proteasome; Nrf1; DDI2; bounce back; proteostasis 1. Introduction Proteins are essential to the vast majority of cellular and organismal functions. Cellular protein levels are defined by the balance among protein synthesis, folding, and degradation, and the failure to accurately coordinate these processes leads to disease [1]. -

Lecture 7: Sequence Motif Discovery
Sequence motif: definitions COSC 348: Computing for Bioinformatics • In Bioinformatics, a sequence motif is a nucleotide or amino-acid sequence pattern that is widespread and has Lecture 7: been proven or assumed to have a biological significance. Sequence Motif Discovery • Once we know the sequence pattern of the motif, then we can use the search methods to find it in the sequences (i.e. Lubica Benuskova Boyer-Moore algorithm, Rabin-Karp, suffix trees, etc.) • The problem is to discover the motifs, i.e. what is the order of letters the particular motif is comprised of. http://www.cs.otago.ac.nz/cosc348/ 1 2 Examples of motifs in DNA Sequence motif: notations • The TATA promoter sequence is an example of a highly • An example of a motif in a protein: N, followed by anything but P, conserved DNA sequence motif found in eukaryotes. followed by either S or T, followed by anything but P − One convention is to write N{P}[ST]{P} where {X} means • Another example of motifs: binding sites for transcription any amino acid except X; and [XYZ] means either X or Y or Z. factors (TF) near promoter regions of genes, etc. • Another notation: each ‘.’ signifies any single AA, and each ‘*’ Gene 1 indicates one member of a closely-related AA family: Gene 2 − WDIND*.*P..*...D.F.*W***.**.IYS**...A.*H*S*WAMRN Gene 3 Gene 4 • In the 1st assignment we have motifs like A??CG, where the Gene 5 wildcard ? Stands for any of A,U,C,G. Binding sites for TF 3 4 Sequence motif discovery from conservation Motif discovery based on alignment • profile analysis is another word for this.