An Invigorating Approach to Free-Radical Reactions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Auxiliary Iron–Sulfur Cofactors in Radical SAM Enzymes☆
Biochimica et Biophysica Acta 1853 (2015) 1316–1334 Contents lists available at ScienceDirect Biochimica et Biophysica Acta journal homepage: www.elsevier.com/locate/bbamcr Review Auxiliary iron–sulfur cofactors in radical SAM enzymes☆ Nicholas D. Lanz a, Squire J. Booker a,b,⁎ a Department of Biochemistry and Molecular Biology, The Pennsylvania State University, University Park, PA 16802, United States b Department of Chemistry, The Pennsylvania State University, University Park, PA 16802, United States article info abstract Article history: A vast number of enzymes are now known to belong to a superfamily known as radical SAM, which all contain a Received 19 September 2014 [4Fe–4S] cluster ligated by three cysteine residues. The remaining, unligated, iron ion of the cluster binds in Received in revised form 15 December 2014 contact with the α-amino and α-carboxylate groups of S-adenosyl-L-methionine (SAM). This binding mode Accepted 6 January 2015 facilitates inner-sphere electron transfer from the reduced form of the cluster into the sulfur atom of SAM, Available online 15 January 2015 resulting in a reductive cleavage of SAM to methionine and a 5′-deoxyadenosyl radical. The 5′-deoxyadenosyl Keywords: radical then abstracts a target substrate hydrogen atom, initiating a wide variety of radical-based transforma- – Radical SAM tions. A subset of radical SAM enzymes contains one or more additional iron sulfur clusters that are required Iron–sulfur cluster for the reactions they catalyze. However, outside of a subset of sulfur insertion reactions, very little is known S-adenosylmethionine about the roles of these additional clusters. This review will highlight the most recent advances in the identifica- Cofactor maturation tion and characterization of radical SAM enzymes that harbor auxiliary iron–sulfur clusters. -

Noncanonical Coproporphyrin-Dependent Bacterial Heme Biosynthesis Pathway That Does Not Use Protoporphyrin
Noncanonical coproporphyrin-dependent bacterial heme biosynthesis pathway that does not use protoporphyrin Harry A. Daileya,b,c,1, Svetlana Gerdesd, Tamara A. Daileya,b,c, Joseph S. Burcha, and John D. Phillipse aBiomedical and Health Sciences Institute and Departments of bMicrobiology and cBiochemistry and Molecular Biology, University of Georgia, Athens, GA 30602; dMathematics and Computer Science Division, Argonne National Laboratory, Argonne, IL 60439; and eDivision of Hematology, Department of Medicine, University of Utah School of Medicine, Salt Lake City, UT 84132 Edited by J. Clark Lagarias, University of California, Davis, CA, and approved January 12, 2015 (received for review August 25, 2014) It has been generally accepted that biosynthesis of protoheme of a “primitive” pathway in Desulfovibrio vulgaris (13). This path- (heme) uses a common set of core metabolic intermediates that way, named the “alternative heme biosynthesis” path (or ahb), has includes protoporphyrin. Herein, we show that the Actinobacteria now been characterized by Warren and coworkers (15) in sulfate- and Firmicutes (high-GC and low-GC Gram-positive bacteria) are reducing bacteria. In the ahb pathway, siroheme, synthesized unable to synthesize protoporphyrin. Instead, they oxidize copro- from uroporphyrinogen III, can be further metabolized by suc- porphyrinogen to coproporphyrin, insert ferrous iron to make Fe- cessive demethylation and decarboxylation to yield protoheme (14, coproporphyrin (coproheme), and then decarboxylate coproheme 15) (Fig. 1 and Fig. S1). A similar pathway exists for protoheme- to generate protoheme. This pathway is specified by three genes containing archaea (15, 16). named hemY, hemH, and hemQ. The analysis of 982 representa- Current gene annotations suggest that all enzymes for pro- tive prokaryotic genomes is consistent with this pathway being karyotic heme synthetic pathways are now identified. -

Sulfur-Containing Radicals: from EPR Spectroscopic Studies to Synthetic Methodology
Sulfur-containing Radicals: From EPR Spectroscopic Studies to Synthetic Methodology A Thesis Presented to the University of London in Partial Fulfilment of the Requirements for the Degree of Doctor of Philosophy Alistair John Fielding February 2002 Christopher Ingold Laboratories Department of Chemistry University College London London WCIH OAJ UCL ProQuest Number: 10015687 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. uest. ProQuest 10015687 Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved. This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC. ProQuest LLC 789 East Eisenhower Parkway P.O. Box 1346 Ann Arbor, Ml 48106-1346 7 believe a leaf of grass is no less than the journey-work of the stars. ” Walt Whitman This thesis is dedicated to my late Auntie, Doreen Latter. 1930-2001 ABSTRACT A variety of novel homochiral silanethiols have been prepared and investigated as polarity-reversal catalysts for the enantioselective hydrosilylation of a prochiral alkene. The enantiomeric excesses of the products were disappointingly small. A computer modelling procedure, based on molecular mechanics, was applied in order to understand the results and quantitative agreement with experiment was reasonably good. Silanethiols have been oxidised to the corresponding disulfides and these have been investigated as photochemical sources of silanethiyl radicals. -

Changes in Soil Microbial Communities After Long-Term Warming Exposure" (2019)
University of Massachusetts Amherst ScholarWorks@UMass Amherst Doctoral Dissertations Dissertations and Theses October 2019 CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG- TERM WARMING EXPOSURE William G. Rodríguez-Reillo University of Massachusetts Amherst Follow this and additional works at: https://scholarworks.umass.edu/dissertations_2 Part of the Environmental Microbiology and Microbial Ecology Commons Recommended Citation Rodríguez-Reillo, William G., "CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE" (2019). Doctoral Dissertations. 1757. https://doi.org/10.7275/15007293 https://scholarworks.umass.edu/dissertations_2/1757 This Open Access Dissertation is brought to you for free and open access by the Dissertations and Theses at ScholarWorks@UMass Amherst. It has been accepted for inclusion in Doctoral Dissertations by an authorized administrator of ScholarWorks@UMass Amherst. For more information, please contact [email protected]. CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE A Dissertation Presented by WILLIAM GABRIEL RODRÍGUEZ-REILLO Submitted to the Graduate School of the University of Massachusetts Amherst in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY SEPTEMBER 2019 Organismic and Evolutionary Biology © Copyright by William Gabriel Rodríguez-Reillo 2019 All Rights Reserved CHANGES IN SOIL MICROBIAL COMMUNITIES AFTER LONG-TERM WARMING EXPOSURE A Dissertation Presented by WILLIAM GABRIEL RODRÍGUEZ-REILLO Approved as to style and content by: _________________________________________ Jeffrey L. Blanchard, Chair _________________________________________ Courtney Babbitt, Member _________________________________________ David Sela, Member _________________________________________ Kristina Stinson, Member ______________________________________ Paige Warren, Graduate Program Director Organismic and Evolutionary Biology DEDICATION To my parents, William Rodriguez Arce and Carmen L. Reillo Batista. A quienes aún en la distancia me mantuvieron en sus oraciones. -

The Crystal Structure of Biotin Synthase, an S-Adenosylmethionine-Dependent Radical Enzyme F
2-74 LIFE SCIENCES SCIENCE HIGHLIGHTS 2-75 The Crystal Structure of Biotin Synthase, an S-Adenosylmethionine-Dependent Radical Enzyme F. Berkovitch1, Y. Nicolet1, J.T. Wan2, J.T. Jarrett2, and C.L. Drennan1 1Department of Chemistry, Massachusetts Institute of Technology; 2Johnson Research Foundation and Department of Biochemistry and Biophysics, University of Pennsylvania BEAMLINE X25 The crystal structure of biotin synthase addresses how “AdoMet radical” enzymes, also called “Radical SAM” enzymes, use an Fe S cluster and S-adenosyl-L-methionine to 4 4 Funding generate organic radicals. Biotin synthase catalyzes the radical-mediated insertion of National Institutes of Health; Searle Scholars Program; sulfur into dethiobiotin (DTB) to form biotin (vitamin B8). The structure places the substrates, i.e. DTB and AdoMet, between the Fe S cluster (essential for radical gen- Cecil and Ida Green Career 4 4 Development Fund; Lester eration) and the Fe2S2 cluster (postulated to be the source of sulfur), with both clusters Wolfe Predoctoral Fellowship; in unprecedented coordination environments. Cellular, Biochemical, and Molecular Sciences training Biotin is an essential vitamin that plays a ubiquitous role in human growth and grant; U.S. Department of Energy; National Institute of metabolism. Biotin deficiency results in skin lesions, abnormal fat distribution, General Medical Sciences neurological symptoms, and immunodeficiency. A low biotin level has also been correlated to an increased incidence of type II diabetes mellitus. Biotin is a valuable Publication commercial commodity, used as an additive in food, health, and cosmetic prod- F. Berkovitch, Y. Nicolet, J.T. Wan, J.T. Jarrett, and ucts, and as a research tool in the biochemical sciences. -

Radical SAM Enzymes in the Biosynthesis of Ribosomally Synthesized and Post-Translationally Modified Peptides (Ripps) Alhosna Benjdia, Clémence Balty, Olivier Berteau
Radical SAM enzymes in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs) Alhosna Benjdia, Clémence Balty, Olivier Berteau To cite this version: Alhosna Benjdia, Clémence Balty, Olivier Berteau. Radical SAM enzymes in the biosynthesis of ribosomally synthesized and post-translationally modified peptides (RiPPs). Frontiers in Chemistry, Frontiers Media, 2017, 5, 10.3389/fchem.2017.00087. hal-02627786 HAL Id: hal-02627786 https://hal.inrae.fr/hal-02627786 Submitted on 26 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License REVIEW published: 08 November 2017 doi: 10.3389/fchem.2017.00087 Radical SAM Enzymes in the Biosynthesis of Ribosomally Synthesized and Post-translationally Modified Peptides (RiPPs) Alhosna Benjdia*, Clémence Balty and Olivier Berteau* Micalis Institute, ChemSyBio, INRA, AgroParisTech, Université Paris-Saclay, Jouy-en-Josas, France Ribosomally-synthesized and post-translationally modified peptides (RiPPs) are a large and diverse family of natural products. They possess interesting biological properties such as antibiotic or anticancer activities, making them attractive for therapeutic applications. In contrast to polyketides and non-ribosomal peptides, RiPPs derive from ribosomal peptides and are post-translationally modified by diverse enzyme families. -

Metabolic Functions of the Human Gut Microbiota: the Role of Metalloenzymes Cite This: DOI: 10.1039/C8np00074c Lauren J
Natural Product Reports View Article Online REVIEW View Journal Metabolic functions of the human gut microbiota: the role of metalloenzymes Cite this: DOI: 10.1039/c8np00074c Lauren J. Rajakovich and Emily P. Balskus * Covering: up to the end of 2017 The human body is composed of an equal number of human and microbial cells. While the microbial community inhabiting the human gastrointestinal tract plays an essential role in host health, these organisms have also been connected to various diseases. Yet, the gut microbial functions that modulate host biology are not well established. In this review, we describe metabolic functions of the human gut microbiota that involve metalloenzymes. These activities enable gut microbial colonization, mediate interactions with the host, and impact human health and disease. We highlight cases in which enzyme characterization has advanced our understanding of the gut microbiota and examples that illustrate the diverse ways in which Received 14th August 2018 Creative Commons Attribution 3.0 Unported Licence. metalloenzymes facilitate both essential and unique functions of this community. Finally, we analyze Human DOI: 10.1039/c8np00074c Microbiome Project sequencing datasets to assess the distribution of a prominent family of metalloenzymes rsc.li/npr in human-associated microbial communities, guiding future enzyme characterization efforts. 1 Introduction our gut from infancy and plays a critical role in the development 2 Commensal colonization of the human gut: tness and and maintenance of healthy human physiology. It aids in adaptation developing the innate and adaptive immune systems,1,2 2.1 Glycan sulfation provides nutrients and vitamins,3 and protects against path- 4 This article is licensed under a 2.2 Glycan fucosylation ogen invasion. -

2018 Summer Symposium Program
Ofce of UNDERGRADUATE RESEARCH ® THE UNIVERSITY OF UTAH 2018 Summer Symposium THURSDAY, AUGUST 2, 2018 9:00AM - 12:00PM CROCKER SCIENCE CENTER UNIVERSITY OF UTAH 2018 SUMMER SYMPOSIUM Thursday, August 2, 2018 9:00 AM – 12:00 PM Crocker Science Center University of Utah The Office of Undergraduate Research is grateful for the generous support of the Office of the Vice President for Research. We are also thankful for the development of the Summer Research Program Partnership, which is a new collaboration among the Chemistry Research Experience for Undergraduates (REU), the Huntsman Cancer Institute’s PathMaker Cancer Research Program, the Native American Summer Research Internship (NARI), the Physics & Astronomy REU and Summer Undergraduate Research Program, and the Summer Program for Undergraduate Research (SPUR). Together, these programs are serving more than 90 undergraduate researchers in Summer 2018. Finally, we would like to express our utmost pride and congratulations to the students, graduate students, and faculty mentors without whose efforts and dedication this event would not be possible. PROGRAM SCHEDULE NOTE: All student presenters MUST check-in Snacks available at 10:15 AM in Room 205/206 8:30 – 9:00 AM CHECK-IN & POSTER SET-UP 9:00 – 10:30 AM POSTER SESSION I (ODD POSTERS) 10:30 AM – 12:00 PM POSTER SESSION II (EVEN POSTERS) 12:00 – 12:15 PM POSTER TAKE-DOWN 2 SCHEDULE OF PRESENTATIONS POSTER SESSION I 9:00 – 10:30 AM Poster 1 Presenter: Sara Alektiar (University of Michigan) Mentor: Matthew Sigman (Chemistry) Electrocatalytic Bis(bipyidine)ruthenium Hydroxylation of Tertiary and Benzylic C-H Bonds The Sigman and Du Bois labs recently reported a methodology that employs a bis(bipyridine)Ru catalyst operating in acidic water to achieve oxidation of tertiary and benzylic C-H bonds in the presence of basic amines. -
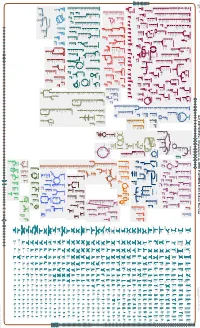
Generated by SRI International Pathway Tools Version 25.0 on Mon
Authors: Pallavi Subhraveti Ron Caspi Peter Midford Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000716605Cyc: Streptomyces sp. NRRL F-525 NRRL F-525 Cellular Overview Connections between pathways are omitted for legibility. Anamika Kothari an amino an amino an amino an amino an amino an amino an amino an amino phosphate phosphate acid acid acid acid acid acid acid acid molybdate an amino an amino H + a dipeptide phosphate phosphate phosphate acid an amino acid an amino H + phosphate phosphate glycine betaine glycine betaine glycine betaine leu pro pro pro acid acid predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted predicted ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC ABC F0F1 ATP ABC RS16420 RS10685 RS06340 RS46495 RS16425 ProP RS28920 RS26410 LeuE RS27350 RS38790 RS13800 transporter transporter transporter RS41005 transporter transporter transporter transporter transporter CoxB transporter transporter transporter synthase transporter of an of an of an of an of an of an of an of an of molybdate of phosphate of phosphate of a dipeptide amino acid -

Notice: Restrictions
NOTICE: The copyright law of the United States (Title 17, United States Code) governs the making of reproductions of copyrighted material. One specified condition is that the reproduction is not to be "used for any purpose other than private study, scholarship, or research." If a user makes a request for, or later uses a reproduction for purposes in excess of "fair use," that user may be liable for copyright infringement. RESTRICTIONS: This student work may be read, quoted from, cited, for purposes of research. It may not be published in full except by permission of the author. Abstract: The phylogenetic position of a number of bacteria within the family Flavobacteriaceae has been questioned. To address the question, the whole genomes of several organisms were sequenced, and this project is focused on Chryseobacterium haifense. The advances in next generation sequencing (NGS) technologies have caused a decrease in cost for whole genome sequencing. This decreased cost has led to more genomes being sequenced and in the process has caused a large demand for bioinformatics tools to handle the genomic data. To analyze the genomic data, the 930,000 reads were assembled in several steps, using several different software packages to refine the assembly to fewer than 700 contiguous sequences. Automated annotation using the Rapid Annotation using Subsystem Technologies (RAST) server identified the organism’s genes and known pathways which were compared to its phenotypes. The Reciprocal Orthology Score Average (ROSA) genomic similarity calculator showed that Chryseobacterium haifense is as different from “true” Chryseobacteria as other separate genera are which has led to the conclusion that Chryseobacterium haifense does not belong within the Chryseobacterium genus. -

Mechanistic Insights Into the Radical S-Adenosyl L-Methionine Enzyme MFTC
University of Denver Digital Commons @ DU Electronic Theses and Dissertations Graduate Studies 1-1-2017 Mechanistic Insights into the Radical S-Adenosyl L-Methionine Enzyme MFTC Bulat Khaliullin University of Denver Follow this and additional works at: https://digitalcommons.du.edu/etd Part of the Biochemistry, Biophysics, and Structural Biology Commons, and the Chemistry Commons Recommended Citation Khaliullin, Bulat, "Mechanistic Insights into the Radical S-Adenosyl L-Methionine Enzyme MFTC" (2017). Electronic Theses and Dissertations. 1300. https://digitalcommons.du.edu/etd/1300 This Thesis is brought to you for free and open access by the Graduate Studies at Digital Commons @ DU. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Digital Commons @ DU. For more information, please contact [email protected],[email protected]. MECHANISTIC INSIGHTS INTO THE RADICAL S-ADENOSYL L-METHIONINE ENZYME MFTC __________ A Thesis Presented to the Faculty of Natural Sciences and Mathematics University of Denver __________ In Partial Fulfillment of the Requirements for the Degree Master of Science __________ by Bulat Khaliullin June 2017 Advisor: John A. Latham ©Copyright by Bulat Khaliullin 2017 All Rights Reserved Author: Bulat Khaliullin Title: MECHANISTIC INSIGHTS INTO THE RADICAL S-ADENOSYL L- METHIONINE ENZYME MFTC Advisor: John A. Latham Degree Date: June 2017 Abstract Mycofactocin is a putative peptide-derived redox cofactor in Mycobacterium family. Its putative biosynthetic pathway is encoded by the operon mftABCDEF. The initial step of this pathway is a posttranslational modification of a peptide precursor MftA, which is catalyzed by MftC enzyme. This modification only occurs in the presence of chaperone MftB. -

Structural Insights Into Recognition and Repair of UV-DNA Damage by Spore Photoproduct Lyase, a Radical SAM Enzyme Alhosna Benjdia1,*, Korbinian Heil2, Thomas R
9308–9318 Nucleic Acids Research, 2012, Vol. 40, No. 18 Published online 2 July 2012 doi:10.1093/nar/gks603 Structural insights into recognition and repair of UV-DNA damage by Spore Photoproduct Lyase, a radical SAM enzyme Alhosna Benjdia1,*, Korbinian Heil2, Thomas R. M. Barends1, Thomas Carell2,* and Ilme Schlichting1,* 1Department of Biomolecular Mechanisms, Max-Planck Institute for Medical Research, Jahnstrasse 29, 69120 Heidelberg and 2Department of Chemistry, Center for Integrated Protein Science (CiPSM), Ludwig-Maximilians University, Butenandtstrasse 5-13, 81377 Munich, Germany Received April 2, 2012; Revised May 26, 2012; Accepted May 29, 2012 ABSTRACT INTRODUCTION Bacterial spores possess an enormous resistance Given the myriad of DNA-damaging agents, bacteria have to ultraviolet (UV) radiation. This is largely due to a evolved diverse DNA repair pathways to resist and survive unique DNA repair enzyme, Spore Photoproduct up to thousands of years in the extreme case of spores. Lyase (SP lyase) that repairs a specific UV-induced This tremendous resistance is achieved by a combination DNA lesion, the spore photoproduct (SP), through of several factors including DNA packing assisted by an unprecedented radical-based mechanism. small acid-soluble spore proteins, dehydration and the Unlike DNA photolyases, SP lyase belongs to the high content of dipicolinic acid (2,6-pyridinedicarboxylic emerging superfamily of radical S-adenosyl-L-me- acid) (1). Under these conditions, ultraviolet (UV) radi- thionine (SAM) enzymes and uses a [4Fe–4S]1+ ation results in the formation of one major DNA lesion, cluster and SAM to initiate the repair reaction. We the so-called spore photoproduct (SP or 5-(R-thyminyl)- report here the first crystal structure of this enig- 5,6-dihydrothymine) (2,3).