Review of Molecular Taxonomy Studies On
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Growing Trend in Application of Age-Stage, Two-Sex Life Table Theory in Diverse Ecologicaland Pest Management Studies
Journal of the Plant Protection Society Volume 5 2018 Plant Protection Society Nepal Review Article GROWING TREND IN APPLICATION OF AGE-STAGE, TWO-SEX LIFE TABLE THEORY IN DIVERSE ECOLOGICALAND PEST MANAGEMENT STUDIES Sabitri Baral1 and Ratna K. Jha1 ABSTRACT Life table is an effective tool for characterizing the demography of an arthropod to understand the phenomenon in pest population development which is a key for developing IPM strategy. Age-stage, two-sex life tables provide comprehensive insights into the stage differentiation of an arthropods, compared with the traditional female age-specific life tables and any other forms of life tables. The age-stage, two-sex life table approach is applied in diverse type of ecological and pest management research. This article is intended to draw attention of Nepalese researchers towards the significance of demographic studies for development of IPM strategy, advancements in application of age-stage, two-sex life table approach and its computer programs. Altogether more than 694 peer reviewed papers and PhD theses have been published since 1988. Out of this, 92 papers were sampled and reviewed for this paper. In such papers, demography of diverse 50 species of arthropods classified under 27 different families belonging to 10 orders have been studied using this theory to measure their fitness in diverse food and environment condition, to assess their consumption, predation or parasitism capacity and to project their population growth in different scenario, to forecast the timing of control based on the stage structure of pest populations and to be used in mass rearing and harvesting of predators and preys under biological control program. -

Diversity and Resource Choice of Flower-Visiting Insects in Relation to Pollen Nutritional Quality and Land Use
Diversity and resource choice of flower-visiting insects in relation to pollen nutritional quality and land use Diversität und Ressourcennutzung Blüten besuchender Insekten in Abhängigkeit von Pollenqualität und Landnutzung Vom Fachbereich Biologie der Technischen Universität Darmstadt zur Erlangung des akademischen Grades eines Doctor rerum naturalium genehmigte Dissertation von Dipl. Biologin Christiane Natalie Weiner aus Köln Berichterstatter (1. Referent): Prof. Dr. Nico Blüthgen Mitberichterstatter (2. Referent): Prof. Dr. Andreas Jürgens Tag der Einreichung: 26.02.2016 Tag der mündlichen Prüfung: 29.04.2016 Darmstadt 2016 D17 2 Ehrenwörtliche Erklärung Ich erkläre hiermit ehrenwörtlich, dass ich die vorliegende Arbeit entsprechend den Regeln guter wissenschaftlicher Praxis selbständig und ohne unzulässige Hilfe Dritter angefertigt habe. Sämtliche aus fremden Quellen direkt oder indirekt übernommene Gedanken sowie sämtliche von Anderen direkt oder indirekt übernommene Daten, Techniken und Materialien sind als solche kenntlich gemacht. Die Arbeit wurde bisher keiner anderen Hochschule zu Prüfungszwecken eingereicht. Osterholz-Scharmbeck, den 24.02.2016 3 4 My doctoral thesis is based on the following manuscripts: Weiner, C.N., Werner, M., Linsenmair, K.-E., Blüthgen, N. (2011): Land-use intensity in grasslands: changes in biodiversity, species composition and specialization in flower-visitor networks. Basic and Applied Ecology 12 (4), 292-299. Weiner, C.N., Werner, M., Linsenmair, K.-E., Blüthgen, N. (2014): Land-use impacts on plant-pollinator networks: interaction strength and specialization predict pollinator declines. Ecology 95, 466–474. Weiner, C.N., Werner, M , Blüthgen, N. (in prep.): Land-use intensification triggers diversity loss in pollination networks: Regional distinctions between three different German bioregions Weiner, C.N., Hilpert, A., Werner, M., Linsenmair, K.-E., Blüthgen, N. -

Evolution of Insect Color Vision: from Spectral Sensitivity to Visual Ecology
EN66CH23_vanderKooi ARjats.cls September 16, 2020 15:11 Annual Review of Entomology Evolution of Insect Color Vision: From Spectral Sensitivity to Visual Ecology Casper J. van der Kooi,1 Doekele G. Stavenga,1 Kentaro Arikawa,2 Gregor Belušic,ˇ 3 and Almut Kelber4 1Faculty of Science and Engineering, University of Groningen, 9700 Groningen, The Netherlands; email: [email protected] 2Department of Evolutionary Studies of Biosystems, SOKENDAI Graduate University for Advanced Studies, Kanagawa 240-0193, Japan 3Department of Biology, Biotechnical Faculty, University of Ljubljana, 1000 Ljubljana, Slovenia; email: [email protected] 4Lund Vision Group, Department of Biology, University of Lund, 22362 Lund, Sweden; email: [email protected] Annu. Rev. Entomol. 2021. 66:23.1–23.28 Keywords The Annual Review of Entomology is online at photoreceptor, compound eye, pigment, visual pigment, behavior, opsin, ento.annualreviews.org anatomy https://doi.org/10.1146/annurev-ento-061720- 071644 Abstract Annu. Rev. Entomol. 2021.66. Downloaded from www.annualreviews.org Copyright © 2021 by Annual Reviews. Color vision is widespread among insects but varies among species, depend- All rights reserved ing on the spectral sensitivities and interplay of the participating photore- Access provided by University of New South Wales on 09/26/20. For personal use only. ceptors. The spectral sensitivity of a photoreceptor is principally determined by the absorption spectrum of the expressed visual pigment, but it can be modified by various optical and electrophysiological factors. For example, screening and filtering pigments, rhabdom waveguide properties, retinal structure, and neural processing all influence the perceived color signal. -

Region of Nymphalidae and Libytheidae (Lepidoptera) from Japan
MORPHOLOGICAL STUDIES ON THE OCCIPITAL REGION OF NYMPHALIDAE AND LIBYTHEIDAE (LEPIDOPTERA) FROM JAPAN Takashi TSUBUKI and Nagao KOYAMA * .h2monji junior High School, 10-33, Kita6tsz{ha 1, Toshima-feu, Toleyo (170), 1opan ** Biological Laboratory, Shinshi2 Udeiversity, Cllada (386), 14pan CONTENTS I. Introduction・--・ny・・・・・・・・・・・・・・・・・-・-・・・・・・-・---・--t・-・---・ny・・・・-・-・・・-・-・・-・・・1 II. Materials and methods・-・・・・・・・・・t・・・・・・t・・・・・・・・・・・・・・`・・・・・・・・・・-・・・-・・・・・・-d・・・・・・・・・・・・2 III. General morphology of the occipital region ・・・・・・}・・・・・・・・・・・・・・・・・・・・・・・・・・・・・4 IV. Occipital structure of each species in Nymphaliclae and Libytheidae ・-ib-・・・・-・・・-・ny・・・・・--・-・・・・・・・-・・-・----・b-・・・----・-・・・-・・-・--・・-6 V. Phylogenical grouping of Nymphalidae based on the occipital structure・t・・・・・・・--・--・・-・-・・・・・・・・・・-・・・・-ny-・・・ny-・--・-----・H"""""".","".."."lg VI. Summary ・・・・-・-・-・・・・・・-・・----・・・-・・・・-・-・-・-----・・-------・-・----・23 VII. Literatures cited ・・・--・・・・・・・・・`・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・・--・-・・-・・・・・・tt・・・・・・23 I. INTRODUCTION A considerable number of studies have been carried out on the occipital morphology of moths since YAGI and KoYAMA's report of 1963 (KoYAMA and MlyATA, 1969, 1970, 1975;MIYATA, 1971, 1972, 1973, 1974, 1975;MIyATA and KOYAMA, 1971, 1972, 1976). In the butterfiies, however, few papers were available concerning the occipital region, on which EHRucH (1958, a b), YAGI and KoyAMA (1963) and KoyAMA and OGAwA (1972) briefly described. Then, tried to study the occipital region of butterflies, the authors observed it preliminarily (TsuBuKI et aL 1975). The present paper deals with the occipital structure of Nymphalidae and Libytheidae from Japan with reference to its bearing on systematics. Before going further, the authors wish to express their gratitude to Prof. Dr. H. SAwADA and Prof. Dr. N. GOKAN, Tokyo University of Agriculture, for their advice and assistance. Thanks are also due to Mr. N. K6DA who gave valuable materials for this work, and to Mr. Y. YAGucm and the members of JEOL Ltd. -

Protophormia Terraenovae (Robineau-Desvoidy, 1830) (Diptera, Calliphoridae) a New Forensic Indicator to South-Western Europe
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Repositorio Institucional de la Universidad de Alicante Ciencia Forense, 12/2015: 137–152 ISSN: 1575-6793 PROTOPHORMIA TERRAENOVAE (ROBINEAU-DESVOIDY, 1830) (DIPTERA, CALLIPHORIDAE) A NEW FORENSIC INDICATOR TO SOUTH-WESTERN EUROPE Anabel Martínez-Sánchez1 Concepción Magaña2 Martin Toniolo Paola Gobbi Santos Rojo Abstract: Protophormia terraenovae larvae are found frequently on corpses in central and northern Europe but are scarce in the Mediterranean area. We present the first case in the Iberian Peninsula where P. terraenovae was captured during autopsies in Madrid (Spain). In the corpse other nec- rophagous flies were found, Lucilia sericata, Chrysomya albiceps and Sarcopha- ga argyrostoma. To calculate the posmortem interval, the life cycle of P. ter- raenovae was studied at constant temperature, room laboratory and natural fluctuating conditions. The total developmental time was 16.61±0.09 days, 16.75±4.99 days in the two first cases. In natural conditions, developmental time varied between 31.22±0.07 days (average temperature: 15.6oC), 15.58±0.08 days (average temperature: 21.5oC) and 14.9±0.10 days (average temperature: 23.5oC). Forensic importance and the implications of other necrophagous Diptera presence is also discussed. Key words: Calliphoridae, forensic entomology, accumulated degrees days, fluctuating temperatures, competition, postmortem interval, Spain. Resumen: Las larvas de Protophormia terraenovae se encuentran con frecuen- cia asociadas a cadáveres en el centro y norte de Europa pero son raras en el área Mediterránea. Presentamos el primer caso en la Península Ibérica don- 1 Departamento de Ciencias Ambientales/Instituto Universitario CIBIO-Centro Iberoame- ricano de la Biodiversidad. -
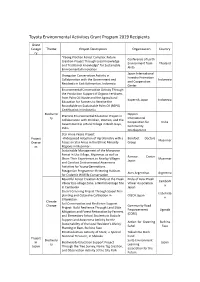
List of Previous Grant Projects
Toyota Environmental Activities Grant Program 2019 Recipients Grant Catego Theme Project Description Organization Country ry "Kaeng Krachan Forest Complex: Future Conference of Earth Creation Project Through Local Knowledge Environment from Thailand and Traditional Knowledge" for Sustainable Akita Environmental Innovation Japan International Orangutan Conservation Activity in Forestry Promotion Collaboration with the Government and Indonesia and Cooperation Residents in East Kalimantan, Indonesia Center Environmental Conservation Activity Through the Production Support of Organic Fertilizers from Palm Oil Waste and the Agricultural Kopernik Japan Indonesia Education for Farmers to Receive the Roundtable on Sustainable Palm Oil (RSPO) Certification in Indonesia Biodiversi Nippon Practical Environmental Education Project in ty International Collaboration with Children, Women, and the Cooperation for India Government in a Rural Village in Bodh Gaya, Community India Development Star Anise Peace Project Project -Widespread Adoption of Agroforestry with a Barefoot Doctors Myanmar Overse Focus on Star Anise in the Ethnic Minority Group as Regions in Myanmar- Sustainable Management of the Mangrove Forest in Uto Village, Myanmar, as well as Ramsar Center Share Their Experiences to Nearby Villages Myanmar Japan and Conduct Environmental Awareness Activities for Young Generations Patagonian Programme: Restoring Habitats Aves Argentinas Argentina for Endemic Wildlife Conservation Beautiful Forest Creation Activity at the Preah Pride of Asia: Preah -

Morphology, Distribution and Abundance of Antennal Sensilla of The
Revista Brasileira de Entomologia 60 (2016) 8–14 REVISTA BRASILEIRA DE Entomologia A Journal on Insect Diversity and Evolution w ww.rbentomologia.com Systematics, Morphology and Biogeography Morphology, distribution and abundance of antennal sensilla of the oyster mushroom fly, Coboldia fuscipes (Meigen) (Diptera: Scatopsidae) a b b,∗ a,∗ b Zhilin Zhang , Xinlian Li , Lizhen Chen , Lihua Wang , Chaoliang Lei a Hubei Key Laboratory of Quality Control of Characteristic Fruits and Vegetables, Hubei Engineering University, Xiaogan, China b Hubei Insect Resources Utilization and Sustainable Pest Management Key Laboratory, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, China a b s t r a c t a r t i c l e i n f o Article history: We investigated the distribution, morphology and abundance of antennae sensilla of Coboldia fuscipes Received 31 May 2015 (Meigen) using scanning electron microscopy. Antennae of C. fuscipes consisted of scape, pedicel, and fla- Accepted 4 November 2015 gellum with eight flagellomeres. Antennal scape and pedicel had only one type of sensillum, i.e., sensilla Available online 26 November 2015 chaetica. Significant differences were found between the number and distribution of these sensilla. Four Associate Editor: Sarah S. Oliveira types of morphologically distinct sensilla on the flagellum were identified, including sensilla chaetica, sensilla trichoidea, sensilla coeloconica, and sensilla basiconica (three subtypes). Significant differences Keywords: were found in the abundance and distribution of sensilla among the antennal flagella and diverse flag- Antennae ellomeres in both sexes. Sensilla trichoidea is the most abundant of sensilla discovered on the antennal Flagellum flagellum. Sensilla chaetica is the largest and longest sensilla among all the types of sensilla found on Scanning electron microscopy the antennal surface of C. -

Phylogeny of Psephenidae (Coleoptera: Byrrhoidea) Based on Larval, Pupal and Adult Characters
Systematic Entomology (2007), 32, 502–538 DOI: 10.1111/j.1365-3113.2006.00374.x Phylogeny of Psephenidae (Coleoptera: Byrrhoidea) based on larval, pupal and adult characters CHI-FENG LEE1 , MASATAKA SATOˆ2 , WILLIAM D. SHEPARD3 and M A N F R E D A . J A¨CH4 1Institute of Biodiversity, National Cheng Kung University, Tainan 701, Taiwan, 2Dia Cuore 306, Kamegahora 3-1404, Midoriku, Nagoya, 458-0804, Japan, 3Essig Museum of Entomology, 201 Wellman Hall, University of California, Berkeley, CA 94720, U.S.A., and 4Natural History Museum, Burgring 7, A-1010 Wien, Austria Abstract. We conducted the first comprehensive phylogenetic analysis of Pse- phenidae, based on 143 morphological characters of adults, larvae and pupae and coded for 34 taxa, representing three outgroups and 31 psephenid genera, including four undescribed ones. A strict consensus tree calculated (439 steps, consistency index ¼ 0.45, retention index ¼ 0.75) from the two most-parsimonious cladograms indicated that the monophyly of the family and subfamilies is supported, with the exception of Eubriinae, which is paraphyletic when including Afroeubria. Here a new subfamily, Afroeubriinae (subfam.n.), is formally estab- lished for Afroeubria. The analysis also indicated that the ‘streamlined’ larva is a derived adaptive radiation. Here, suprageneric taxonomy and the evolution of some significant characters are discussed. Keys are provided to the subfamilies and genera of Psephenidae considering larvae, adults and pupae. Introduction type specimens and the association of adults and immature stages by rearing, five new genera were proposed for Ori- Psephenidae, the ‘water penny’ beetles, are characterized by ental taxa and a well-resolved phylogenetic tree was ob- the peculiar larval body shape (Figs 4–7). -

The Taxonomy of the Japanese Oak Red Scale Insect
Zootaxa 3630 (2): 291–307 ISSN 1175-5326 (print edition) www.mapress.com/zootaxa/ Article ZOOTAXA Copyright © 2013 Magnolia Press ISSN 1175-5334 (online edition) http://dx.doi.org/10.11646/zootaxa.3630.2.5 http://zoobank.org/urn:lsid:zoobank.org:pub:373402BA-36A9-4323-AE9D-A84D89C47231 The taxonomy of the Japanese oak red scale insect, Kuwania quercus (Kuwana) (Hemiptera: Coccoidea: Kuwaniidae), with a generic diagnosis, a key to species and description of a new species from California SAN’AN WU1, NAN NAN1, PENNY GULLAN2 & JUN DENG1 1The Key Laboratory for Silviculture and Conservation of Ministry of Education, Beijing Forestry University, Beijing, P. R. China 100083. E-mail: [email protected] 2Division of Evolution, Ecology and Genetics, Research School of Biology, The Australian National University, Canberra, A.C.T. 0200, Australia. E-mail: [email protected] Abstract The oak red scale insect, Kuwania quercus (Kuwana), was described from specimens collected from the bark of oak trees (Quercus species) in Japan. More recently, the species has been identified from California and China, but Californian specimens differ morphologically from Japanese material and are considered here to be a new species based on both morphological and molecular data. In this paper, an illustrated redescription of K. quercus is provided based on type specimens consisting of adult females, first-instar nymphs and intermediate-stage females, and a lectotype is designated for Sasakia quercus Kuwana. The new Californian species, Kuwania raygilli Wu & Gullan, is described and illustrated based on the adult female, first-instar nymph and intermediate-stage female. A new generic diagnosis for Kuwania Cockerell based on adult females and first-instar nymphs, and a key to species based on adult females are included. -

Terry Whitworth 3707 96Th ST E, Tacoma, WA 98446
Terry Whitworth 3707 96th ST E, Tacoma, WA 98446 Washington State University E-mail: [email protected] or [email protected] Published in Proceedings of the Entomological Society of Washington Vol. 108 (3), 2006, pp 689–725 Websites blowflies.net and birdblowfly.com KEYS TO THE GENERA AND SPECIES OF BLOW FLIES (DIPTERA: CALLIPHORIDAE) OF AMERICA, NORTH OF MEXICO UPDATES AND EDITS AS OF SPRING 2017 Table of Contents Abstract .......................................................................................................................... 3 Introduction .................................................................................................................... 3 Materials and Methods ................................................................................................... 5 Separating families ....................................................................................................... 10 Key to subfamilies and genera of Calliphoridae ........................................................... 13 See Table 1 for page number for each species Table 1. Species in order they are discussed and comparison of names used in the current paper with names used by Hall (1948). Whitworth (2006) Hall (1948) Page Number Calliphorinae (18 species) .......................................................................................... 16 Bellardia bayeri Onesia townsendi ................................................... 18 Bellardia vulgaris Onesia bisetosa ..................................................... -

Molecular Analysis of Forensically Important Blow Flies in Thailand
insects Article Molecular Analysis of Forensically Important Blow Flies in Thailand Narin Sontigun 1, Kabkaew L. Sukontason 1, Jens Amendt 2, Barbara K. Zajac 3, Richard Zehner 2, Kom Sukontason 1, Theeraphap Chareonviriyaphap 4 and Anchalee Wannasan 1,* 1 Department of Parasitology, Faculty of Medicine, Chiang Mai University, Chiang Mai 50200, Thailand; [email protected] (N.S.); [email protected] (K.L.S.); [email protected] (K.S.) 2 Institute of Legal Medicine, Forensic Biology/Entomology, Kennedyallee 104, Frankfurt am Main 60596, Germany; [email protected] (J.A.); [email protected] (R.Z.) 3 Department for Forensic Genetics, Institute of Forensic Medicine and Traffic Medicine, Voßstraße 2, Heidelberg 69115, Germany; [email protected] 4 Department of Entomology, Faculty of Agriculture, Kasetsart University, Bangkok 10900, Thailand; [email protected] * Correspondence: [email protected]; Tel.: +66-8-9434-6851 Received: 13 September 2018; Accepted: 6 November 2018; Published: 8 November 2018 Abstract: Blow flies are the first insect group to colonize on a dead body and thus correct species identification is a crucial step in forensic investigations for estimating the minimum postmortem interval, as developmental times are species-specific. Due to the difficulty of traditional morphology-based identification such as the morphological similarity of closely related species and uncovered taxonomic keys for all developmental stages, DNA-based identification has been increasing in interest, especially