Keratins and Skin Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Development and Maintenance of Epidermal Stem Cells in Skin Adnexa
International Journal of Molecular Sciences Review Development and Maintenance of Epidermal Stem Cells in Skin Adnexa Jaroslav Mokry * and Rishikaysh Pisal Medical Faculty, Charles University, 500 03 Hradec Kralove, Czech Republic; [email protected] * Correspondence: [email protected] Received: 30 October 2020; Accepted: 18 December 2020; Published: 20 December 2020 Abstract: The skin surface is modified by numerous appendages. These structures arise from epithelial stem cells (SCs) through the induction of epidermal placodes as a result of local signalling interplay with mesenchymal cells based on the Wnt–(Dkk4)–Eda–Shh cascade. Slight modifications of the cascade, with the participation of antagonistic signalling, decide whether multipotent epidermal SCs develop in interfollicular epidermis, scales, hair/feather follicles, nails or skin glands. This review describes the roles of epidermal SCs in the development of skin adnexa and interfollicular epidermis, as well as their maintenance. Each skin structure arises from distinct pools of epidermal SCs that are harboured in specific but different niches that control SC behaviour. Such relationships explain differences in marker and gene expression patterns between particular SC subsets. The activity of well-compartmentalized epidermal SCs is orchestrated with that of other skin cells not only along the hair cycle but also in the course of skin regeneration following injury. This review highlights several membrane markers, cytoplasmic proteins and transcription factors associated with epidermal SCs. Keywords: stem cell; epidermal placode; skin adnexa; signalling; hair pigmentation; markers; keratins 1. Epidermal Stem Cells as Units of Development 1.1. Development of the Epidermis and Placode Formation The embryonic skin at very early stages of development is covered by a surface ectoderm that is a precursor to the epidermis and its multiple derivatives. -

Bilateral Lower Extremity Hyperkeratotic Plaques: a Case Report of Ichthyosis Vulgaris
Faculty & Staff Scholarship 2015 Bilateral lower extremity hyperkeratotic plaques: a case report of ichthyosis vulgaris Hayley Leight Zachary Zinn Omid Jalali Follow this and additional works at: https://researchrepository.wvu.edu/faculty_publications Clinical, Cosmetic and Investigational Dermatology Dovepress open access to scientific and medical research Open Access Full Text Article CASE REPORT Bilateral lower extremity hyperkeratotic plaques: a case report of ichthyosis vulgaris Hayley Leight Abstract: Here, we report a case of a middle-aged woman presenting with severe, long-standing, Zachary Zinn hyperkeratotic plaques of the lower extremities unrelieved by over-the-counter medications. Omid Jalali Initial history and clinical findings were suggestive of an inherited ichthyosis. Ichthyoses are genetic disorders characterized by dry scaly skin and altered skin-barrier function. A diagnosis Department of Dermatology, West Virginia University, of ichthyosis vulgaris was confirmed by histopathology. Etiology, prevalence, and treatment Morgantown, WV, USA options are discussed. Keywords: filaggrin gene, FLG, profilaggrin, keratohyalin granules, hyperkeratosis Introduction For personal use only. Inherited ichthyoses are a diverse group of genetic disorders characterized by dry, scaly skin; hyperkeratosis; and altered skin-barrier function. While these disorders of cutaneous keratinization are multifaceted and varying in etiology, disruption in the stratum corneum with generalized scaling is common to all.1–4 Although not entirely known -

Associated Palmoplantar Keratoderma
DR ABIGAIL ZIEMAN (Orcid ID : 0000-0001-8236-207X) Article type : Review Article Pathophysiology of pachyonychia congenita-associated palmoplantar keratoderma: New insight into skin epithelial homeostasis and avenues for treatment Authors: A. G. Zieman1 and P. A. Coulombe1,2 # Affiliations: 1Department of Cell and Developmental Biology, University of Michigan Medical School, Ann Arbor, MI 48109, USA; 2Department of Dermatology, University of Michigan Medical School, Ann Arbor, MI 48109, USA #Corresponding author: Pierre A. Coulombe, PhD, 3071 Biomedical Sciences Research Building, 109 Zina Pitcher Place, Ann Arbor, MI 48109, USA. Tel: 734-615-7509. Email: [email protected]. Funding Sources: These studies were supported by grant AR044232 issued to P.A.C. from the National Institute of Arthritis, Musculoskeletal and Skin Disease (NIAMS). A.G.Z. received support from grant T32 CA009110 from the National Cancer Institute. Author Manuscript This is the author manuscript accepted for publication and has undergone full peer review but has not been through the copyediting, typesetting, pagination and proofreading process, which may lead to differences between this version and the Version of Record. Please cite this article as doi: 10.1111/BJD.18033 This article is protected by copyright. All rights reserved Conflict of interest disclosures: None declared. Bulleted statements: What’s already known about this topic? Pachyonychia congenita is a rare genodermatosis caused by mutations in KRT6A, KRT6B, KRT6C, KRT16, KRT17, which are normally expressed in skin appendages and induced following injury. Individuals with PC present with multiple clinical symptoms that usually include thickened and dystrophic nails, palmoplantar keratoderma (PPK), glandular cysts, and oral leukokeratosis. -

Studies on the Proteome of Human Hair - Identifcation of Histones and Deamidated Keratins Received: 15 August 2017 Sunil S
www.nature.com/scientificreports OPEN Studies on the Proteome of Human Hair - Identifcation of Histones and Deamidated Keratins Received: 15 August 2017 Sunil S. Adav 1, Roopa S. Subbaiaih2, Swat Kim Kerk 2, Amelia Yilin Lee 2,3, Hui Ying Lai3,4, Accepted: 12 January 2018 Kee Woei Ng3,4,7, Siu Kwan Sze 1 & Artur Schmidtchen2,5,6 Published: xx xx xxxx Human hair is laminar-fbrous tissue and an evolutionarily old keratinization product of follicle trichocytes. Studies on the hair proteome can give new insights into hair function and lead to the development of novel biomarkers for hair in health and disease. Human hair proteins were extracted by detergent and detergent-free techniques. We adopted a shotgun proteomics approach, which demonstrated a large extractability and variety of hair proteins after detergent extraction. We found an enrichment of keratin, keratin-associated proteins (KAPs), and intermediate flament proteins, which were part of protein networks associated with response to stress, innate immunity, epidermis development, and the hair cycle. Our analysis also revealed a signifcant deamidation of keratin type I and II, and KAPs. The hair shafts were found to contain several types of histones, which are well known to exert antimicrobial activity. Analysis of the hair proteome, particularly its composition, protein abundances, deamidated hair proteins, and modifcation sites, may ofer a novel approach to explore potential biomarkers of hair health quality, hair diseases, and aging. Hair is an important and evolutionarily conserved structure. It originates from hair follicles deep within the der- mis and is mainly composed of hair keratins and KAPs, which form a complex network that contributes to the rigidity and mechanical properties. -

Molecular Pathways Associated with the Nutritional Programming of Plant-Based Diet Acceptance in Rainbow Trout Following an Early Feeding Exposure Mukundh N
Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure Mukundh N. Balasubramanian, Stéphane Panserat, Mathilde Dupont-Nivet, Edwige Quillet, Jérôme Montfort, Aurélie Le Cam, Françoise Médale, Sadasivam J. Kaushik, Inge Geurden To cite this version: Mukundh N. Balasubramanian, Stéphane Panserat, Mathilde Dupont-Nivet, Edwige Quillet, Jérôme Montfort, et al.. Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure. BMC Genomics, BioMed Central, 2016, 17 (1), pp.1-20. 10.1186/s12864-016-2804-1. hal-01346912 HAL Id: hal-01346912 https://hal.archives-ouvertes.fr/hal-01346912 Submitted on 19 Jul 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Balasubramanian et al. BMC Genomics (2016) 17:449 DOI 10.1186/s12864-016-2804-1 RESEARCH ARTICLE Open Access Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure Mukundh N. Balasubramanian1, Stephane Panserat1, Mathilde Dupont-Nivet2, Edwige Quillet2, Jerome Montfort3, Aurelie Le Cam3, Francoise Medale1, Sadasivam J. Kaushik1 and Inge Geurden1* Abstract Background: The achievement of sustainable feeding practices in aquaculture by reducing the reliance on wild-captured fish, via replacement of fish-based feed with plant-based feed, is impeded by the poor growth response seen in fish fed high levels of plant ingredients. -
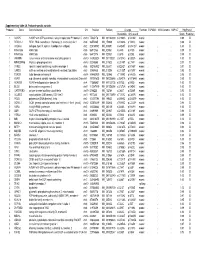
Supplementary Table 2A. Proband-Specific Variants Proband
Supplementary table 2A. Proband-specific variants Proband Gene Gene full name Chr Position RefSeqChange Function ESP6500 1000 Genome SNP137 PolyPhen2 Nucleotide Amino acid Score Prediction 1 AGAP5 ArfGAP with GTPase domain, ankyrin repeat and PH domain 5 chr10 75435178 NM_001144000 c.G1240A p.V414M exonic . 0.99 D 1 REXO1L1 REX1, RNA exonuclease 1 homolog (S. cerevisiae)-like 1 chr8 86574465 NM_172239 c.A1262G p.Y421C exonic . 0.98 D 1 COL4A3 collagen, type IV, alpha 3 (Goodpasture antigen) chr2 228169782 NM_000091 c.G4235T p.G1412V exonic . 1.00 D 1 KIAA1586 KIAA1586 chr6 56912133 NM_020931 c.C44G p.A15G exonic . 0.99 D 1 KIAA1586 KIAA1586 chr6 56912176 NM_020931 c.C87G p.D29E exonic . 0.99 D 1 JAKMIP3 Janus kinase and microtubule interacting protein 3 chr10 133955524 NM_001105521 c.G1574C p.G525A exonic . 1.00 D 1 MPHOSPH8 M-phase phosphoprotein 8 chr13 20240685 NM_017520 c.C2140T p.L714F exonic . 0.97 D 1 SYNE1 spectrin repeat containing, nuclear envelope 1 chr6 152783902 NM_033071 c.A2242T p.N748Y exonic . 0.97 D 1 CAND2 cullin-associated and neddylation-dissociated 2 (putative) chr3 12858835 NM_012298 c.C2125T p.H709Y exonic . 0.95 D 1 TDRD9 tudor domain containing 9 chr14 104460909 NM_153046 c.T1289G p.V430G exonic . 0.96 D 2 ASPM asp (abnormal spindle) homolog, microcephaly associated (Drosophila)chr1 197091625 NM_001206846 c.G3491A p.R1164H exonic . 0.99 D 2 ADAM29 ADAM metallopeptidase domain 29 chr4 175896951 NM_001130705 c.A275G p.D92G exonic . 1.00 D 2 BCO2 beta-carotene oxygenase 2 chr11 112087019 NM_001256398 c.G1373A p.G458E exonic . -

Hypomesus Transpacificus
Aquatic Toxicology 105 (2011) 369–377 Contents lists available at ScienceDirect Aquatic Toxicology jou rnal homepage: www.elsevier.com/locate/aquatox Sublethal responses to ammonia exposure in the endangered delta smelt; Hypomesus transpacificus (Fam. Osmeridae) ∗ 1 2 Richard E. Connon , Linda A. Deanovic, Erika B. Fritsch, Leandro S. D’Abronzo , Inge Werner Aquatic Toxicology Laboratory, Department of Anatomy, Physiology and Cell Biology, School of Veterinary Medicine, University of California, Davis, California 95616, United States a r t i c l e i n f o a b s t r a c t Article history: The delta smelt (Hypomesus transpacificus) is an endangered pelagic fish species endemic to the Received 9 May 2011 Sacramento-San Joaquin Estuary in Northern California, which acts as an indicator of ecosystem health Received in revised form 29 June 2011 in its habitat range. Interrogative tools are required to successfully monitor effects of contaminants upon Accepted 2 July 2011 the delta smelt, and to research potential causes of population decline in this species. We used microarray technology to investigate genome-wide effects in fish exposed to ammonia; one of multiple contami- Keywords: nants arising from wastewater treatment plants and agricultural runoff. A 4-day exposure of 57-day Hypomesus transpacificus + old juveniles resulted in a total ammonium (NH4 –N) median lethal concentration (LC50) of 13 mg/L, Delta smelt Microarray and a corresponding un-ionized ammonia (NH3) LC50 of 147 g/L. Using the previously designed delta + Biomarker smelt microarray we assessed altered gene transcription in juveniles exposed to 10 mg/L NH4 –N from Ammonia this 4-day exposure. -

Keratin 9 Point Mutation in the Pedigree of Epidermolytic Hereditary Palmoplantar Keratoderma Perturbs Keratin Intermediate Filament Network Formation
FEBS 17004 FEBS Letters 386 (1996) 149-155 Keratin 9 point mutation in the pedigree of epidermolytic hereditary palmoplantar keratoderma perturbs keratin intermediate filament network formation Setsu Kobayashi, Toshihiro Tanaka*, Norihisa Matsuyoshi, Sadao Imamura Department of Dermatology, Graduate School of Medicine, Kyoto University, Kyoto, 606 Japan Received 12 January 1996; revised version received 4 April 1996 Abstract Keratins form an intracellular keratin filament net- point mutations in the K9 gene in EHPPK [4-8] but none work in keratinocytes. Point mutations in the epidermal keratins showed a function assay with these mutations. Here, we pro- could lead to the disruption of keratin filament formation, vide the first demonstration that the point mutation found in developing skin diseases such as epidermolytic hereditary a pedigree of EHPPK has a dominant-negative effect on the palmoplantar keratoderma (EHPPK). We found a G to A assembly of keratin intermediate filaments in the cells. transition in keratin 9 (K9) cDNA, resulting in the substitution of glutamine for arginine at 162, in all patients of a pedigree of 2. Materials and methods EHPPK. Transfection into MDCK cells and DJM-1 cells revealed that the plasmid CMX vector containing normal keratin 2.1. PCR and DNA sequence 9 cDNA showed normal keratin network formation, whereas the Genomic DNA was extracted and purified from blood or biopsy vector with a G to A point mutated keratin 9 cDNA showed specimens from the patients. The primers were designed at nucleotide disrupted keratin filaments with droplet formation in the cells. 263 282 and 664-683 based on the K9 cDNA sequence [9]. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Structural and Biochemical Changes Underlying a Keratoderma-Like Phenotype in Mice Lacking Suprabasal AP1 Transcription Factor Function
Citation: Cell Death and Disease (2015) 6, e1647; doi:10.1038/cddis.2015.21 OPEN & 2015 Macmillan Publishers Limited All rights reserved 2041-4889/15 www.nature.com/cddis Structural and biochemical changes underlying a keratoderma-like phenotype in mice lacking suprabasal AP1 transcription factor function EA Rorke*,1, G Adhikary2, CA Young2, RH Rice3, PM Elias4, D Crumrine4, J Meyer4, M Blumenberg5 and RL Eckert2,6,7,8 Epidermal keratinocyte differentiation on the body surface is a carefully choreographed process that leads to assembly of a barrier that is essential for life. Perturbation of keratinocyte differentiation leads to disease. Activator protein 1 (AP1) transcription factors are key controllers of this process. We have shown that inhibiting AP1 transcription factor activity in the suprabasal murine epidermis, by expression of dominant-negative c-jun (TAM67), produces a phenotype type that resembles human keratoderma. However, little is understood regarding the structural and molecular changes that drive this phenotype. In the present study we show that TAM67-positive epidermis displays altered cornified envelope, filaggrin-type keratohyalin granule, keratin filament, desmosome formation and lamellar body secretion leading to reduced barrier integrity. To understand the molecular changes underlying this process, we performed proteomic and RNA array analysis. Proteomic study of the corneocyte cross-linked proteome reveals a reduction in incorporation of cutaneous keratins, filaggrin, filaggrin2, late cornified envelope precursor proteins, hair keratins and hair keratin-associated proteins. This is coupled with increased incorporation of desmosome linker, small proline-rich, S100, transglutaminase and inflammation-associated proteins. Incorporation of most cutaneous keratins (Krt1, Krt5 and Krt10) is reduced, but incorporation of hyperproliferation-associated epidermal keratins (Krt6a, Krt6b and Krt16) is increased. -

CDH12 Cadherin 12, Type 2 N-Cadherin 2 RPL5 Ribosomal
5 6 6 5 . 4 2 1 1 1 2 4 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 A A A A A A A A A A A A A A A A A A A A C C C C C C C C C C C C C C C C C C C C R R R R R R R R R R R R R R R R R R R R B , B B B B B B B B B B B B B B B B B B B , 9 , , , , 4 , , 3 0 , , , , , , , , 6 2 , , 5 , 0 8 6 4 , 7 5 7 0 2 8 9 1 3 3 3 1 1 7 5 0 4 1 4 0 7 1 0 2 0 6 7 8 0 2 5 7 8 0 3 8 5 4 9 0 1 0 8 8 3 5 6 7 4 7 9 5 2 1 1 8 2 2 1 7 9 6 2 1 7 1 1 0 4 5 3 5 8 9 1 0 0 4 2 5 0 8 1 4 1 6 9 0 0 6 3 6 9 1 0 9 0 3 8 1 3 5 6 3 6 0 4 2 6 1 0 1 2 1 9 9 7 9 5 7 1 5 8 9 8 8 2 1 9 9 1 1 1 9 6 9 8 9 7 8 4 5 8 8 6 4 8 1 1 2 8 6 2 7 9 8 3 5 4 3 2 1 7 9 5 3 1 3 2 1 2 9 5 1 1 1 1 1 1 5 9 5 3 2 6 3 4 1 3 1 1 4 1 4 1 7 1 3 4 3 2 7 6 4 2 7 2 1 2 1 5 1 6 3 5 6 1 3 6 4 7 1 6 5 1 1 4 1 6 1 7 6 4 7 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m -

Ichthyosis Hystrix
Case Report Ichthyosis hystrix Surajit Nayak, Basanti Acharjya, Prasenjit Mohanty Department of Skin ABSTRACT and VD, MKCG Medical College and Hospital, The present report describes the condition in a three day old male child with bilateral ,linear, hyperpigmented and Berhampur, Orissa, India hyperkeratotic verrucous plaques and patchy alopecia over scalpe without any nail and skeletal abnormalities. It was suggestive of ichthyosis hystrix type of epidermal nevus,and is being reported in view of the rarity of this condition. Key words: Icthyosis hystrix, epidermal nevus syndrome, etretinate INTRODUCTION most part of the face. Nails were normal. In the lower limbs, in addition to the nevus, there were Ichthyosis hystrix the nomenclature comes unilateral hyperpigmented [Figure 1] macular from the Greek word and condition was first patches encircling right upper thigh and complete described in England in early 18th century. left thigh, sparing a band-like zone. The term ichthyosis hystrix is used to describe several rare skin disorders in the ichthyosis On physical examination, we could not observe family of skin disorders characterized by massive any defects, especially in skeletal or central hyperkeratosis with an appearance like spiny nervous systems. Routine laboratory examination scales. The term has also been employed to including complete blood count, urine analysis, describe localized and linear warty epidermal nevi liver function test and chest X-ray were all within sometimes associated with mental retardation, normal limits. The parents did not permit a biopsy. seizures or skeletal anomalies. Alopecia and hair and nail abnormalities as well as inner ear Based on the above constellation of clinical deafness were also seen in these patients.