A Novel Nonsense Variant of the AGXT Identified in a Chinese Family
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CDH12 Cadherin 12, Type 2 N-Cadherin 2 RPL5 Ribosomal
5 6 6 5 . 4 2 1 1 1 2 4 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 A A A A A A A A A A A A A A A A A A A A C C C C C C C C C C C C C C C C C C C C R R R R R R R R R R R R R R R R R R R R B , B B B B B B B B B B B B B B B B B B B , 9 , , , , 4 , , 3 0 , , , , , , , , 6 2 , , 5 , 0 8 6 4 , 7 5 7 0 2 8 9 1 3 3 3 1 1 7 5 0 4 1 4 0 7 1 0 2 0 6 7 8 0 2 5 7 8 0 3 8 5 4 9 0 1 0 8 8 3 5 6 7 4 7 9 5 2 1 1 8 2 2 1 7 9 6 2 1 7 1 1 0 4 5 3 5 8 9 1 0 0 4 2 5 0 8 1 4 1 6 9 0 0 6 3 6 9 1 0 9 0 3 8 1 3 5 6 3 6 0 4 2 6 1 0 1 2 1 9 9 7 9 5 7 1 5 8 9 8 8 2 1 9 9 1 1 1 9 6 9 8 9 7 8 4 5 8 8 6 4 8 1 1 2 8 6 2 7 9 8 3 5 4 3 2 1 7 9 5 3 1 3 2 1 2 9 5 1 1 1 1 1 1 5 9 5 3 2 6 3 4 1 3 1 1 4 1 4 1 7 1 3 4 3 2 7 6 4 2 7 2 1 2 1 5 1 6 3 5 6 1 3 6 4 7 1 6 5 1 1 4 1 6 1 7 6 4 7 e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e e l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l l p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p p m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m m -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -

AMPK Signaling Regulates Expression of Urea Cycle Enzymes in Response to Changes in Dietary Protein Intake
bioRxiv preprint doi: https://doi.org/10.1101/439380; this version posted October 10, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. AMPK signaling regulates expression of urea cycle enzymes in response to changes in dietary protein intake Sandra Kirsch Heibel1Y, Peter J McGuire2Y, Nantaporn Haskins1, Himani Datta Majumdar1, Sree Rayavarapu1‡, Kanneboyina Nagaraju3, Yetrib Hathout3, Kristy Brown1, Mendel Tuchman1, Ljubica Caldovic1*, 1 Center for Genetic Medicine Research/Children's National Medical Center, Washington, DC, USA 2 National Human Genome Research Institute/National Institutes for Health, Bethesda, MD, USA 3 Department of Pharmaceutical Sciences/Binghamton University, Binghamton NY, USA YThese authors contributed equally to this work. ‡Current address: Division of Clinical Review, Office of Bioequivalence, Office of Generic Drugs, Center for Drug Evaluation and Research/Food and Drug Administration, Silver Spring, MD, USA * [email protected] Abstract Abundance of urea cycle enzymes in the liver is regulated by the dietary protein intake. Although urea cycle enzyme levels rise in response to a high protein diet, signaling networks that sense dietary protein intake and trigger changes in expression of urea cycle genes have not been identified. The aim of this study was to identify signaling pathway(s) that respond to changes in protein intake and regulate expression of urea cycle genes in mice and human hepatocytes. Mice were adapted to either control or high (HP) protein diets followed by isolation of liver protein and mRNA and integrated analysis of the proteomic and transcriptome profiles. -

AGXT Gene Mutations and Their Influence on Clinical Heterogeneity of Type 1 Primary Hyperoxaluria
J Am Soc Nephrol 12: 2072–2079, 2001 AGXT Gene Mutations and Their Influence on Clinical Heterogeneity of Type 1 Primary Hyperoxaluria ANTONIO AMOROSO,*† DOROTI PIRULLI,* FIORELLA FLORIAN,‡ DANIELA PUZZER,† MICHELE BONIOTTO,* SERGIO CROVELLA,* SILVIA ZEZLINA,† ANDREA SPANO` ,† GINA MAZZOLA,§ SILVANA SAVOLDI, CRISTINA FERRETTINI,¶ SILVIA BERUTTI,¶ MICHELE PETRARULO,¶ and MARTINO MARANGELLA¶ *Section of Genetics, Department of Reproductive and Developmental Science, University of Trieste, Trieste, Italy; †Medical Genetics Service, IRCCS Burlo Garofolo, Trieste, Italy; ‡Department of Biology, University of Trieste, Trieste, Italy; §Transplant Immunology Service, Ospedale S. Giovanni Battista di Torino, Torino, Italy; Division of Nephrology and Dialysis, Azienda Ospedaliera Triestina, Trieste, Italy; and ¶Renal Stones Center, Ospedale Mauriziano Umberto I, Torino, Italy. Abstract. Primary hyperoxaluria type 1 (PH1) is an autosomal were performed. Both mutant alleles were found in 21 out of recessive disorder that is caused by a deficiency of alanine: 23 patients, and 13 different mutations were recognized in glyoxylate aminotransferase (AGT), which is encoded by a exons 1, 2, 4, and 10. Normalized AGT activity was lower in single copy gene (AGXT). Molecular diagnosis was used in the severe form than in the adult form (P Ͻ 0.05). Double conjunction with clinical, biochemical, and enzymological data heterozygous patients presented a lower age at the onset of the to evaluate genotype-phenotype correlation. Twenty-three un- disease (P ϭ 0.025), and they were more frequent in group A related, Italian PH1 patients were studied, 20 of which were (75%) than in the group B (14%; P ϭ 0.0406). The T444C grouped according to severe form of PH1 (group A), adult mutation was more frequent in the severe form (P Ͻ 0.05), and form (group B), and mild to moderate decrease in renal func- the opposite was observed for G630A (P Ͻ 0.05). -

Primary Hyperoxaluria Type 1: AGT Mistargeting Highlights the Fundamental Differences Between the Peroxisomal and Mitochondrial Protein Import Pathways
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Biochimica et Biophysica Acta 1763 (2006) 1776–1784 www.elsevier.com/locate/bbamcr Review Primary hyperoxaluria type 1: AGT mistargeting highlights the fundamental differences between the peroxisomal and mitochondrial protein import pathways Christopher J. Danpure Department of Biology, University College London, Gower Street, London WC1E 6BT, UK Received 28 April 2006; received in revised form 1 August 2006; accepted 18 August 2006 Available online 24 August 2006 Abstract Primary hyperoxaluria type 1 (PH1) is an atypical peroxisomal disorder, as befits a deficiency of alanine:glyoxylate aminotransferase (AGT), which is itself an atypical peroxisomal enzyme. PH1 is characterized by excessive synthesis and excretion of the metabolic end-product oxalate and the progressive accumulation of insoluble calcium oxalate in the kidney and urinary tract. Disease in many patients is caused by a unique protein trafficking defect in which AGT is mistargeted from peroxisomes to mitochondria, where it is metabolically ineffectual, despite remaining catalytically active. Although the peroxisomal import of human AGT is dependent upon the PTS1 import receptor PEX5p, its PTS1 is exquisitely specific for mammalian AGT, suggesting the presence of additional peroxisomal targeting information elsewhere in the AGT molecule. This and many other functional peculiarities of AGT are probably a consequence of its rather chequered evolutionary history, during which much of its time has been spent being a mitochondrial, rather than a peroxisomal, enzyme. Analysis of the molecular basis of AGT mistargeting in PH1 has thrown into sharp relief some of the fundamental differences between the requirements of the peroxisomal and mitochondrial protein import pathways, particularly the properties of peroxisomal and mitochondrial matrix targeting sequences and the different conformational limitations placed upon importable cargos. -
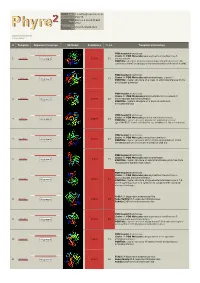
Phyre 2 Results for P50277
Email [email protected] Description P50277 Mon Jul 2 19:25:55 BST Date 2012 Unique Job 2972c4295ed43821 ID Detailed template information # Template Alignment Coverage 3D Model Confidence % i.d. Template Information PDB header:transferase Chain: B: PDB Molecule:adenosylmethionine-8-amino-7- 1 c4a0rB_ Alignment 100.0 23 oxononanoate PDBTitle: structure of bifunctional dapa aminotransferase-dtb synthetase from2 arabidopsis thaliana bound to dethiobiotin (dtb). PDB header:transferase Chain: A: PDB Molecule:aminotransferase, class iii; 2 c3hmuA_ 100.0 27 Alignment PDBTitle: crystal structure of a class iii aminotransferase from2 silicibacter pomeroyi PDB header:transferase Chain: D: PDB Molecule:adenosylmethionine-8-amino-7- 3 c3n5mD_ Alignment 100.0 28 oxononanoate aminotransferase; PDBTitle: crystals structure of a bacillus anthracis aminotransferase PDB header:transferase Chain: A: PDB Molecule:putative aminotransferase; 4 c3fcrA_ 100.0 24 Alignment PDBTitle: crystal structure of putative aminotransferase (yp_614685.1) from2 silicibacter sp. tm1040 at 1.80 a resolution PDB header:transferase Chain: A: PDB Molecule:omega transaminase; 5 c4a6tA_ 100.0 27 Alignment PDBTitle: crystal structure of the omega transaminase from2 chromobacterium violaceum in complex with plp PDB header:transferase Chain: B: PDB Molecule:aminotransferase; 6 c3i5tB_ 100.0 25 Alignment PDBTitle: crystal structure of aminotransferase prk07036 from rhodobacter2 sphaeroides kd131 PDB header:transferase Chain: A: PDB Molecule:adenosylmethionine-8-amino-7- oxononanoate -

(12) Patent Application Publication (10) Pub. No.: US 2003/0198970 A1 Roberts (43) Pub
US 2003O19897OA1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2003/0198970 A1 Roberts (43) Pub. Date: Oct. 23, 2003 (54) GENOSTICS clinical trials on groups or cohorts of patients. This group data is used to derive a Standardised method of treatment (75) Inventor: Gareth Wyn Roberts, Cambs (GB) which is Subsequently applied on an individual basis. There is considerable evidence that a significant factor underlying Correspondence Address: the individual variability in response to disease, therapy and FINNEGAN, HENDERSON, FARABOW, prognosis lies in a person's genetic make-up. There have GARRETT & DUNNER been numerous examples relating that polymorphisms LLP within a given gene can alter the functionality of the protein 1300 ISTREET, NW encoded by that gene thus leading to a variable physiological WASHINGTON, DC 20005 (US) response. In order to bring about the integration of genomics into medical practice and enable design and building of a (73) Assignee: GENOSTIC PHARMA LIMITED technology platform which will enable the everyday practice (21) Appl. No.: 10/206,568 of molecular medicine a way must be invented for the DNA Sequence data to be aligned with the identification of genes (22) Filed: Jul. 29, 2002 central to the induction, development, progression and out come of disease or physiological States of interest. Accord Related U.S. Application Data ing to the invention, the number of genes and their configu rations (mutations and polymorphisms) needed to be (63) Continuation of application No. 09/325,123, filed on identified in order to provide critical clinical information Jun. 3, 1999, now abandoned. concerning individual prognosis is considerably less than the 100,000 thought to comprise the human genome. -

Metabolic Network-Based Stratification of Hepatocellular Carcinoma Reveals Three Distinct Tumor Subtypes
Metabolic network-based stratification of hepatocellular carcinoma reveals three distinct tumor subtypes Gholamreza Bidkhoria,b,1, Rui Benfeitasa,1, Martina Klevstigc,d, Cheng Zhanga, Jens Nielsene, Mathias Uhlena, Jan Borenc,d, and Adil Mardinoglua,b,e,2 aScience for Life Laboratory, KTH Royal Institute of Technology, SE-17121 Stockholm, Sweden; bCentre for Host-Microbiome Interactions, Dental Institute, King’s College London, SE1 9RT London, United Kingdom; cDepartment of Molecular and Clinical Medicine, University of Gothenburg, SE-41345 Gothenburg, Sweden; dThe Wallenberg Laboratory, Sahlgrenska University Hospital, SE-41345 Gothenburg, Sweden; and eDepartment of Biology and Biological Engineering, Chalmers University of Technology, SE-41296 Gothenburg, Sweden Edited by Sang Yup Lee, Korea Advanced Institute of Science and Technology, Daejeon, Republic of Korea, and approved November 1, 2018 (received for review April 27, 2018) Hepatocellular carcinoma (HCC) is one of the most frequent forms of of markers associated with recurrence and poor prognosis (13–15). liver cancer, and effective treatment methods are limited due to Moreover, genome-scale metabolic models (GEMs), collections tumor heterogeneity. There is a great need for comprehensive of biochemical reactions, and associated enzymes and transporters approaches to stratify HCC patients, gain biological insights into have been successfully used to characterize the metabolism of subtypes, and ultimately identify effective therapeutic targets. We HCC, as well as identify drug targets for HCC patients (11, 16–18). stratified HCC patients and characterized each subtype using tran- For instance, HCC tumors have been stratified based on the uti- scriptomics data, genome-scale metabolic networks and network lization of acetate (11). Analysis of HCC metabolism has also led topology/controllability analysis. -

Alanine and Aspartate Aminotransferase and Glutamine-Cycling Pathway: Their Roles in Pathogenesis of Metabolic Syndrome
Online Submissions: http://www.wjgnet.com/1007-9327office World J Gastroenterol 2012 August 7; 18(29): 3775-3781 [email protected] ISSN 1007-9327 (print) ISSN 2219-2840 (online) doi:10.3748/wjg.v18.i29.3775 © 2012 Baishideng. All rights reserved. FIELD OF VISION Alanine and aspartate aminotransferase and glutamine-cycling pathway: Their roles in pathogenesis of metabolic syndrome Silvia Sookoian, Carlos J Pirola Silvia Sookoian, Department of Clinical and Molecular Hepa- factors such as obesity, insulin resistance (IR), high tology, Institute of Medical Research A Lanari-IDIM, Univer- blood pressure, and dyslipidemia were associated with sity of Buenos Aires-National Council of Scientific and Tech- several metabolites, including branched-chain amino nological Research (CONICET), Ciudad Autónoma de Buenos acids, other hydrophobic amino acids, tryptophan Aires 1427, Argentina breakdown products, and nucleotide metabolites. In Carlos J Pirola, Department of Molecular Genetics and Biol- addition, the authors found a significant association of ogy of Complex Diseases, Institute of Medical Research A IR traits with glutamine, glutamate and the glutamine- Lanari-IDIM, University of Buenos Aires-National Council of Scientific and Technological Research (CONICET), Ciudad Au- to-glutamate ratio. These data provide new insight into tónoma de Buenos Aires 1427, Argentina the pathogenesis of MS-associated phenotypes and in- Author contributions: Sookoian S and Pirola CJ designed the troduce a crucial role of glutamine-cycling pathway as study, analyzed -

Recombinant Production of Eight Human
Recombinant production of eight human cytosolic aminotransferases and assessment of their potential involvement in glyoxylate metabolism Stefano Donini, Manuela Ferrari, Chiara Fedeli, Marco Faini, Ilaria Lamberto, Ada Serena Marletta, Lara Mellini, Michela Panini, Riccardo Percudani, Loredano Pollegioni, et al. To cite this version: Stefano Donini, Manuela Ferrari, Chiara Fedeli, Marco Faini, Ilaria Lamberto, et al.. Recombinant production of eight human cytosolic aminotransferases and assessment of their potential involve- ment in glyoxylate metabolism. Biochemical Journal, Portland Press, 2009, 422 (2), pp.265-272. 10.1042/BJ20090748. hal-00479195 HAL Id: hal-00479195 https://hal.archives-ouvertes.fr/hal-00479195 Submitted on 30 Apr 2010 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Biochemical Journal Immediate Publication. Published on 23 Jun 2009 as manuscript BJ20090748 Recombinant production of eight human cytosolic aminotransferases and assessment of their potential involvement in glyoxylate metabolism. Stefano Donini *, Manuela Ferrari *, Chiara Fedeli *, Marco Faini *‡ , Ilaria Lamberto *, Ada Serena Marletta *, Lara Mellini *, Michela Panini *, Riccardo Percudani *, Loredano Pollegioni †, Laura Caldinelli †, Stefania Petrucco *1 and Alessio Peracchi *2 * Department of Biochemistry and Molecular Biology, University of Parma, 43100 Parma, Italy † The Protein Factory - Department of Biotechnology and Molecular Sciences, University of Insubria, 21100 Varese, Italy. -

AGXT Gene Alanine--Glyoxylate and Serine--Pyruvate Aminotransferase
AGXT gene alanine--glyoxylate and serine--pyruvate aminotransferase Normal Function The AGXT gene provides instructions for making an enzyme called serine-pyruvate aminotransferase. This enzyme is found in liver cells, specifically within cell structures called peroxisomes. These structures are important for several cellular activities, such as ridding the cell of toxic substances and helping to break down certain fats. In the peroxisome, serine-pyruvate aminotransferase converts a compound called glyoxylate to the protein building block (amino acid) glycine. Health Conditions Related to Genetic Changes Primary hyperoxaluria More than 175 mutations in the AGXT gene have been found to cause primary hyperoxaluria type 1. This condition is caused by the overproduction of a substance called oxalate. Excess amounts of this substance lead to kidney and bladder stones, which can begin anytime from childhood to early adulthood with kidney disease developing at any age. Deposition of oxalate in multiple other tissues throughout the body (systemic oxalosis) can cause additional health problems. Most of the AGXT gene mutations decrease or eliminate serine-pyruvate aminotransferase activity, which impairs the conversion of glyoxylate to glycine. Other mutations cause the enzyme to be misplaced in cells, transporting it to structures called mitochondria instead of to peroxisomes. While the enzyme in the mitochondria retains activity, it cannot access glyoxylate, which is in peroxisomes. All AGXT gene mutations result in the accumulation of glyoxylate, which is converted to oxalate instead of glycine. The oxalate is filtered through the kidneys and is either excreted in urine as a waste product or combines with calcium to form calcium oxalate, a hard compound that is the main component of kidney and bladder stones. -

Mutational Analysis of AGXT in Two Chinese Families with Primary
Li et al. BMC Nephrology 2014, 15:92 http://www.biomedcentral.com/1471-2369/15/92 RESEARCH ARTICLE Open Access Mutational analysis of AGXT in two Chinese families with primary hyperoxaluria type 1 Guo-min Li1, Hong Xu1*, Qian Shen1, Yi-nv Gong1, Xiao-yan Fang1, Li Sun1, Hai-mei Liu1 and Yu An2* Abstract Background: Primary hyperoxaluria type 1 is a rare autosomal recessive disease of glyoxylate metabolism caused by a defect in the liver-specific peroxisomal enzyme alanine:glyoxylate aminotransferase (AGT) that leads to hyperoxaluria, recurrent urolithiasis, and nephrocalcinosis. Methods: Two unrelated patients with recurrent urolithiasis, along with members of their families, exhibited mutations in the AGXT gene by PCR direct sequencing. Results: Two heterozygous mutations that predict truncated proteins, p.S81X and p.S275delinsRAfs, were identified in one patient. The p.S81X mutation is novel. Two heterozygous missense mutations, p.M1T and p.I202N, were detected in another patient but were not identified in her sibling. These four mutations were confirmed to be of paternal and maternal origin. Conclusions: These are the first cases of primary hyperoxaluria type 1 to be diagnosed by clinical manifestations and AGXT gene mutations in mainland China. The novel p.S81X and p.I202N mutations detected in our study extend the spectrum of known AGXT gene mutations. Keywords: AGXT gene, Chinese children, Mutational analysis, Novel mutation, Primary hyperoxaluria type 1 Background [5,7-9]. Excessive oxalate excretion is an indicator of Primary hyperoxaluria type 1 (PH1; MIM# 259900) is an this disease; however, the test is not specific for PH1 autosomal recessive disorder of glyoxylate metabolism, and may have misleading results because oxalate excretion leading to the overproduction of endogenous oxalate; may be reduced during renal failure [2,10,11].