Full Text (PDF)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Role of Pak2 Promoting Intrinsic Tumor Cell Motility and Worsening Patient Outcomes in Non-Small Cell Lung Cancer
ROLE OF PAK2 PROMOTING INTRINSIC TUMOR CELL MOTILITY AND WORSENING PATIENT OUTCOMES IN NON-SMALL CELL LUNG CANCER By Adam Marc Bissonnette Thesis Submitted to the Faculty of the Graduate School of Vanderbilt University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE in Cancer Biology May 11, 2018 Nashville, Tennessee Approved: David P. Carbone, M.D., Ph.D. Barbara Fingleton, Ph.D. Vito Quaranta, M.D. To my daughter, Harper, whose eyes, smile, and laugh fill my heart, and My beautiful wife, Sarah, for remaining patient and supportive throughout this endeavor. ii ACKNOWLEDGEMENTS First and foremost, I would like to thank my advisor Dr. David Carbone for providing me all the necessary resources to carry out my research between Vanderbilt University and Ohio State University. Though I may not have always deserved his support as a graduate student, Dr. Carbone always stood firmly in my corner until the very end even pledging his support for my PhD candidacy after a long period of time with little progress towards the completion of my doctoral requirements. For that, I could not be any more grateful. I am also appreciative of Dr. Barbara Fingleton and Dr. Vito Quaranta, for their work on my dissertation committee, as well as the sharing of their precious time to offer me guidance throughout my graduate studies. To all of the students, post-docs, and researchers who I have collaborated with and had the good fortune to cross paths with along this journey of mine, all of these experiences allowed me to respect the passage that was graduate school. -

1 Decision of the Election Committee on a Due Impartiality Complaint Brought by the Respect Party in Relation to the London Deba
Decision of the Election Committee on a due impartiality complaint brought by the Respect Party in relation to The London Debate ITV London, 5 April 2016 LBC 97.3 , 5 April 2016 1. On Friday 29 April 2016, Ofcom’s Election Committee (“the Committee”)1 met to consider and adjudicate on a complaint made by the Respect Party in relation to its candidate for the London Mayoral election, George Galloway (“the Complaint”). The Complaint was about the programme The London Debate, broadcast in ITV’s London region on ITV, and on ITV HD and ITV+1 at 18:00 on Tuesday 5 April 2016 (“the Programme”). The Programme was broadcast simultaneously by LBC on the local analogue radio station LBC 97.3, as well as nationally on DAB radio and on digital television (as a radio channel). 2. The Committee consisted of the following members: Nick Pollard (Chair, Member of the Ofcom Content Board); Dame Lynne Brindley DBE (Member of the Ofcom Board and Content Board); Janey Walker (Member of the Ofcom Content Board); and Tony Close (Ofcom Director with responsibility for Content Standards, Licensing and Enforcement and Member of the Ofcom Content Board). 3. For the reasons set out in this decision, having considered all of the submissions and evidence before it under the relevant provisions of the Broadcasting Code (“the Code”), the Committee decided not to uphold the Respect Party’s complaint. The Committee found that in respect of ITV the broadcast of the Programme complied with the requirements of the Code. In the case of LBC, the Programme did not a contain list of candidates in the 2016 London Mayoral election (in audio form) and LBC therefore breached Rule 6.11. -
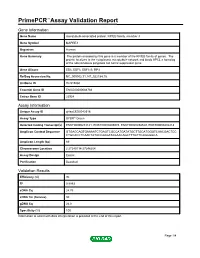
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name microtubule-associated protein, RP/EB family, member 3 Gene Symbol MAPRE3 Organism Human Gene Summary The protein encoded by this gene is a member of the RP/EB family of genes. The protein localizes to the cytoplasmic microtubule network and binds APCL a homolog of the adenomatous polyposis coli tumor suppressor gene. Gene Aliases EB3, EBF3, EBF3-S, RP3 RefSeq Accession No. NC_000002.11, NT_022184.15 UniGene ID Hs.515860 Ensembl Gene ID ENSG00000084764 Entrez Gene ID 22924 Assay Information Unique Assay ID qHsaCED0042518 Assay Type SYBR® Green Detected Coding Transcript(s) ENST00000233121, ENST00000405074, ENST00000458529, ENST00000402218 Amplicon Context Sequence GTGACCAGTGAAAATCTGAGTCGCCATGATATGCTTGCATGGGTCAACGACTCC CTGCACCTCAACTATACCAAGATAGAACAGCTTTGTTCAGGGGCA Amplicon Length (bp) 69 Chromosome Location 2:27245114-27246204 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 90 R2 0.9993 cDNA Cq 24.76 cDNA Tm (Celsius) 80 gDNA Cq 26.8 Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 1/4 PrimePCR™Assay Validation Report MAPRE3, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification Standard Curve Standard curve generated using 20 million copies of template diluted 10-fold to 20 copies Page 2/4 PrimePCR™Assay Validation Report Products used to generate validation data Real-Time PCR Instrument CFX384 Real-Time PCR Detection System Reverse Transcription Reagent iScript™ Advanced cDNA Synthesis Kit for RT-qPCR Real-Time PCR Supermix SsoAdvanced™ SYBR® Green Supermix Experimental Sample qPCR Human Reference Total RNA Data Interpretation Unique Assay ID This is a unique identifier that can be used to identify the assay in the literature and online. -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

Cells RI-Dependent Signal Transduction in Mast Ε Rhoh Plays
RhoH Plays Critical Roles in Fcε RI-Dependent Signal Transduction in Mast Cells This information is current as Hiroyo Oda, Manabu Fujimoto, Michael S. Patrick, Dai of October 2, 2021. Chida, Yoshinori Sato, Yoshinao Azuma, Hiroki Aoki, Takaya Abe, Harumi Suzuki and Mutsunori Shirai J Immunol 2009; 182:957-962; ; doi: 10.4049/jimmunol.182.2.957 http://www.jimmunol.org/content/182/2/957 Downloaded from References This article cites 45 articles, 13 of which you can access for free at: http://www.jimmunol.org/content/182/2/957.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on October 2, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2009 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology RhoH Plays Critical Roles in FcRI-Dependent Signal Transduction in Mast Cells1 Hiroyo Oda,* Manabu Fujimoto,† Michael S. Patrick,*‡ Dai Chida,* Yoshinori Sato,* Yoshinao Azuma,‡ Hiroki Aoki,§ Takaya Abe,¶ Harumi Suzuki,2* and Mutsunori Shirai‡ RhoH is an atypical small G protein with defective GTPase activity that is specifically expressed in hematopoietic lineage cells. -

A Rac-Cgmp Signaling Pathway
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector A Rac-cGMP Signaling Pathway Dagang Guo,1 Ying-cai Tan,1 Dawei Wang,1 K.S. Madhusoodanan,1 Yi Zheng,2 Thomas Maack,1 J. Jillian Zhang,1 and Xin-Yun Huang1,* 1 Department of Physiology, Cornell University Weill Medical College, New York, NY 10021, USA 2 Division of Experimental Hematology, Children’s Hospital Research Foundation, University of Cincinnati, Cincinnati, OH 45229, USA *Correspondence: [email protected] DOI 10.1016/j.cell.2006.11.048 SUMMARY et al., 2004). PAK family members regulate cellular prolif- eration, differentiation, transformation, and survival. They The small GTPase Rac and the second messen- also play important roles in cytoskeleton rearrangement ger cGMP (guanosine 30,50-cyclic monophos- during cell migration. Expression of constitutively active phate) are critical regulators of diverse cell PAK stimulates ruffle formation and inhibits stress fibers functions. When activated by extracellular (Manser et al., 1997; Sells et al., 1997). Increases in PAK signals via membrane signaling receptors, Rac expression and activity have been correlated with pro- executes its functions through engaging down- gression of colorectal carcinomas to metastasis (Carter et al., 2004) and enhanced motility and invasiveness of stream effectors such as p21-activated kinase human breast cancer cells (Vadlamudi et al., 2000). In (PAK), a serine/threonine protein kinase. How- mammals, PAKs can be grouped into two subfamilies: ever, the molecular mechanism by which mem- group A (PAK1, 2, and 3) can be activated by small brane signaling receptors regulate cGMP levels GTPases such as Rac-GTP or Cdc42-GTP binding is not known. -

Expression of Rho Gtpases Family in Melanoma Cells and Its Influence on Cytoskeleton and Migration
www.impactjournals.com/oncotarget/ Oncotarget, 2017, Vol. 8, (No. 18), pp: 30112-30122 Research Paper Expression of Rho GTPases family in melanoma cells and its influence on cytoskeleton and migration Si-Jian Wen1,2, Wei Zhang2, Na-Na Ni3, Qiong Wu2, Xiao-Po Wang2, You-Kun Lin1 Jian-Fang Sun2 1Department of Dermatology and Venerology, First Affiliated Hospital of Guangxi Medical University, Nanning 530021, China 2Department of Pathology, Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, Nanjing 210042, China 3Jiangsu Key Laboratory of Molecular Biology for Skin Diseases and STIs, Nanjing 210042, China Correspondence to: You-Kun Lin, email: [email protected] Jian-Fang Sun, email: [email protected] Keywords: Rho GTPase, melanoma, cytoskeleton, cell migration Received: November 01, 2016 Accepted: January 11, 2017 Published: February 22, 2017 Copyright: Wen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC-BY), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. ABSTRACT Rho GTPases family members influenced the filopodia, lamellipodia, stress fiber and adhesion plaque of melanoma cells through regulating cytoskeleton recombination. The role of Rho GTPases family in the migration and invasion of melanoma and its molecular mechanism were explored. The morphological difference between three types of melanoma cells (M14, A375 and MV3) and human melanocyte (MC) was observed by the Hoffman microscope. Cells were stained by phalloidin labeled by rhodamine. The differences between 4 types of cells in filopodia, lamellipodia, stress fiber and adhesion plaque (microfilament is the main constituent) were observed under the super-high resolution microscope. -
HCC and Cancer Mutated Genes Summarized in the Literature Gene Symbol Gene Name References*
HCC and cancer mutated genes summarized in the literature Gene symbol Gene name References* A2M Alpha-2-macroglobulin (4) ABL1 c-abl oncogene 1, receptor tyrosine kinase (4,5,22) ACBD7 Acyl-Coenzyme A binding domain containing 7 (23) ACTL6A Actin-like 6A (4,5) ACTL6B Actin-like 6B (4) ACVR1B Activin A receptor, type IB (21,22) ACVR2A Activin A receptor, type IIA (4,21) ADAM10 ADAM metallopeptidase domain 10 (5) ADAMTS9 ADAM metallopeptidase with thrombospondin type 1 motif, 9 (4) ADCY2 Adenylate cyclase 2 (brain) (26) AJUBA Ajuba LIM protein (21) AKAP9 A kinase (PRKA) anchor protein (yotiao) 9 (4) Akt AKT serine/threonine kinase (28) AKT1 v-akt murine thymoma viral oncogene homolog 1 (5,21,22) AKT2 v-akt murine thymoma viral oncogene homolog 2 (4) ALB Albumin (4) ALK Anaplastic lymphoma receptor tyrosine kinase (22) AMPH Amphiphysin (24) ANK3 Ankyrin 3, node of Ranvier (ankyrin G) (4) ANKRD12 Ankyrin repeat domain 12 (4) ANO1 Anoctamin 1, calcium activated chloride channel (4) APC Adenomatous polyposis coli (4,5,21,22,25,28) APOB Apolipoprotein B [including Ag(x) antigen] (4) AR Androgen receptor (5,21-23) ARAP1 ArfGAP with RhoGAP domain, ankyrin repeat and PH domain 1 (4) ARHGAP35 Rho GTPase activating protein 35 (21) ARID1A AT rich interactive domain 1A (SWI-like) (4,5,21,22,24,25,27,28) ARID1B AT rich interactive domain 1B (SWI1-like) (4,5,22) ARID2 AT rich interactive domain 2 (ARID, RFX-like) (4,5,22,24,25,27,28) ARID4A AT rich interactive domain 4A (RBP1-like) (28) ARID5B AT rich interactive domain 5B (MRF1-like) (21) ASPM Asp (abnormal -

Appendix 1 – Press Monitoring (National Publications)
APPENDIX 1 – PRESS MONITORING (NATIONAL PUBLICATIONS) Sunday Daily Nationals Magazines Nationals Other Daily Express Anglers Mail Daily Star City AM London Sunday Daily Mail Angling Times Sunday Times Lloyds List London Daily Star CIWM Magazine The Mail on International Sunday Herald Tribune Daily Telegraph Economist News of the Wall Street World Journal Europe Evening Standard ENDS Report The Observer Financial Times Farmers Guardian Sunday Express Metro Farmers Weekly Sunday Mirror The Guardian The People Independent i Local Government Chronicle Independent Materials Recycling Week Daily Mirror Motorboats Monthly The Sun Municipal Journal The Times Regeneration & Renewal New Statesman New Scientist PR Week Planning Private Eye Recycling and Waste World Utility Week Resource Management and Recovery Reuters New Civil Engineer APPENDIX 2 – PRESS MONITORING (NATIONAL KEY WORDS) Keyword Keyword Description Agriculture - Environment Important mentions of farming or agriculture ICW the environment. Carbon Emissions All mentions of carbon emissions OICW climate change, global warming etc. Climate Change All mentions of climate change. Coastal Erosion All mentions of coastal erosion. DEFRA All mentions of Department for Environment, Food and Rural Affairs (DEFRA). Department of Energy & Climate All mentions of the Department of Energy & Climate Change Change (DECC) Drought Main focus mentions of droughts in the UK Environment All mentions of environmental issues Environment - Emissions Important mentions of emissions and their effect on the environment. Environment Agency All mentions of the Environment Agency. Fishing All mentions of fishing ICW the environment. Flooding - Environmental Impact Important Mentions of floods OICW effects on the environment and homes Fly Tipping All mentions of fly tipping Hosepipes All mentions of hosepipes Nuclear Power - Environmental Main focus mentions of environmental effects of nuclear power. -

EB3 (MAPRE3) (NM 012326) Human Tagged ORF Clone Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for RC204135 EB3 (MAPRE3) (NM_012326) Human Tagged ORF Clone Product data: Product Type: Expression Plasmids Product Name: EB3 (MAPRE3) (NM_012326) Human Tagged ORF Clone Tag: Myc-DDK Symbol: MAPRE3 Synonyms: EB3; EBF3; EBF3-S; RP3 Vector: pCMV6-Entry (PS100001) E. coli Selection: Kanamycin (25 ug/mL) Cell Selection: Neomycin ORF Nucleotide >RC204135 ORF sequence Sequence: Red=Cloning site Blue=ORF Green=Tags(s) TTTTGTAATACGACTCACTATAGGGCGGCCGGGAATTCGTCGACTGGATCCGGTACCGAGGAGATCTGCC GCCGCGATCGCC ATGGCCGTCAATGTGTACTCCACATCTGTGACCAGTGAAAATCTGAGTCGCCATGATATGCTTGCATGGG TCAACGACTCCCTGCACCTCAACTATACCAAGATAGAACAGCTTTGTTCAGGGGCAGCCTACTGCCAGTT CATGGACATGCTCTTCCCCGGCTGTGTGCACTTGAGGAAAGTGAAGTTCCAGGCCAAACTAGAGCATGAA TACATCCACAACTTCAAGGTGCTGCAAGCAGCTTTCAAGAAGATGGGTGTTGACAAAATCATTCCTGTAG AGAAATTAGTGAAAGGAAAATTCCAAGATAATTTTGAGTTTATTCAGTGGTTTAAGAAATTCTTTGACGC AAACTATGATGGAAAGGATTACAACCCTCTGCTGGCGCGGCAGGGCCAGGACGTAGCGCCACCTCCTAAC CCAGGTGATCAGATCTTCAACAAATCCAAGAAACTCATTGGCACAGCAGTTCCACAGAGGACGTCCCCCA CAGGCCCAAAAAACATGCAGACCTCTGGCCGGCTGAGCAATGTGGCCCCCCCCTGCATTCTCCGGAAGAA TCCTCCATCAGCCCGAAATGGCGGCCATGAGACTGATGCCCAAATTCTTGAACTCAACCAACAGCTGGTG GACTTGAAGCTGACAGTGGATGGGCTGGAGAAGGAACGTGACTTCTACTTCAGCAAACTTCGTGACATCG AGCTCATCTGCCAGGAGCATGAAAGTGAAAACAGCCCTGTTATCTCAGGCATCATTGGCATCCTCTATGC CACAGAGGAAGGATTCGCACCCCCTGAGGACGATGAGATTGAAGAGCATCAACAAGAAGACCAGGACGAG TAC ACGCGTACGCGGCCGCTCGAGCAGAAACTCATCTCAGAAGAGGATCTGGCAGCAAATGATATCCTGGATT -

Supporting Information
Supporting Information Pouryahya et al. SI Text Table S1 presents genes with the highest absolute value of Ricci curvature. We expect these genes to have significant contribution to the network’s robustness. Notably, the top two genes are TP53 (tumor protein 53) and YWHAG gene. TP53, also known as p53, it is a well known tumor suppressor gene known as the "guardian of the genome“ given the essential role it plays in genetic stability and prevention of cancer formation (1, 2). Mutations in this gene play a role in all stages of malignant transformation including tumor initiation, promotion, aggressiveness, and metastasis (3). Mutations of this gene are present in more than 50% of human cancers, making it the most common genetic event in human cancer (4, 5). Namely, p53 mutations play roles in leukemia, breast cancer, CNS cancers, and lung cancers, among many others (6–9). The YWHAG gene encodes the 14-3-3 protein gamma, a member of the 14-3-3 family proteins which are involved in many biological processes including signal transduction regulation, cell cycle pro- gression, apoptosis, cell adhesion and migration (10, 11). Notably, increased expression of 14-3-3 family proteins, including protein gamma, have been observed in a number of human cancers including lung and colorectal cancers, among others, suggesting a potential role as tumor oncogenes (12, 13). Furthermore, there is evidence that loss Fig. S1. The histogram of scalar Ricci curvature of 8240 genes. Most of the genes have negative scalar Ricci curvature (75%). TP53 and YWHAG have notably low of p53 function may result in upregulation of 14-3-3γ in lung cancer Ricci curvatures. -

G Protein-Coupled Receptors
G PROTEIN-COUPLED RECEPTORS Overview:- The completion of the Human Genome Project allowed the identification of a large family of proteins with a common motif of seven groups of 20-24 hydrophobic amino acids arranged as α-helices. Approximately 800 of these seven transmembrane (7TM) receptors have been identified of which over 300 are non-olfactory receptors (see Frederikson et al., 2003; Lagerstrom and Schioth, 2008). Subdivision on the basis of sequence homology allows the definition of rhodopsin, secretin, adhesion, glutamate and Frizzled receptor families. NC-IUPHAR recognizes Classes A, B, and C, which equate to the rhodopsin, secretin, and glutamate receptor families. The nomenclature of 7TM receptors is commonly used interchangeably with G protein-coupled receptors (GPCR), although the former nomenclature recognises signalling of 7TM receptors through pathways not involving G proteins. For example, adiponectin and membrane progestin receptors have some sequence homology to 7TM receptors but signal independently of G-proteins and appear to reside in membranes in an inverted fashion compared to conventional GPCR. Additionally, the NPR-C natriuretic peptide receptor has a single transmembrane domain structure, but appears to couple to G proteins to generate cellular responses. The 300+ non-olfactory GPCR are the targets for the majority of drugs in clinical usage (Overington et al., 2006), although only a minority of these receptors are exploited therapeutically. Signalling through GPCR is enacted by the activation of heterotrimeric GTP-binding proteins (G proteins), made up of α, β and γ subunits, where the α and βγ subunits are responsible for signalling. The α subunit (tabulated below) allows definition of one series of signalling cascades and allows grouping of GPCRs to suggest common cellular, tissue and behavioural responses.