Molybdenum-99 (99Mo): Past, Present, and Future
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

High Accuracy Measurement of Isotope Ratios of Molybdenum in Some Terrestrial Molybdenites
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector ARTICLES High Accuracy Measurement of Isotope Ratios of Molybdenum in Some Terrestrial Molybdenites Qi-Lu* and Akimasa Masuda Department of Chemistry, Faculty of Science, The University of Tokyo, Tokyo, Japan The isotope ratios of molybdenum in molybdenites were studied. A special triple filament technique was used to obtain stable and lasting signals for MO+. There are no differences bigger than ~0.4 parts per IO4 among four samples and the standard. CJ Am Sot Mass Spectrom 1992, 3, IO- 17) olybdenum is a very interesting element be- denum thus far, in spite of the potential importance cause its seven isotopes can reflect several of research in isotopic abundance of molybdenum. M effects related to nuclear physics. The nu- In this study we have established a method for clear phenomena that may affect the isotope ratios in securing stable and lasting current of MO+ and exam- question are (1) the synthesis of seven isotopes of MO ined the mass fractionation of MO isotopes during involving three processes (r, s, and p) in the standard measurement. Based on these studies, the isotope model of nucleosynthesis [I]; ($2 the nuclear hssion of ratios of MO were determined with high precision uranium-238, which produces MO, “MO, 98Mo, and for some molybdenites from a variety of locations ‘“MO; and (3) the double-beta decays of ‘“Zr and throughout the world. The present study will afford a lwMo leading to 96Mo and ‘“Ru. Another intriguing foundation for further precise studies of molybdenum property of this element is that the anomalous abun- isotopes involving meteorites and terrestrial rocks. -

Isotope Production Potential at Sandia National Laboratories: Product, Waste, Packaging, and Transportation*
Isotope Production Potential at Sandia National Laboratories: Product, Waste, Packaging, and Transportation* A. J. Trennel Transportation Systems Department *- *-, o / /"-~~> Sandia National Laboratories** ' J Albuquerque, NM 87185 O Q T » Abstract The U.S. Congress directed the U.S. Department of Energy to establish a domestic source of molybdenum-99, an essential isotope used in nuclear medicine and radiopharmacology. An Environmental Impact Statement for production of 99Mo at one of four candidate sites is being prepared. As one of the candidate sites, Sandia National Laboratories is developing the Isotope Production Project. Using federally approved processes and procedures now owned by the U.S. Department of Energy, and existing facilities that would be modified to meet the production requirements, the Sandia National Laboratories' Isotope Project would manufacture up to 30 percent of the U.S. market, with the capacity to meet 100 percent of the domestic need if necessary. This paper provides a brief overview of the facility, equipment, and processes required to produce isotopes. Packaging and transportation issues affecting both product and waste are addressed, and the storage and disposal of the four low-level radioactive waste types generated by the production program are considered. Recommendations for future development are provided. This work was performed at Sandia National Laboratories, Albuquerque, New Mexico, for the U.S. Department of Energy under Contract DE-AC04-94AL85000. A U.S. Department of Energy facility. DISTRPJTO OF THIS DOCUMENT IS UNLIMITED #t/f W A8 1 fcll PROJECT NEED AND BACKGROUND Nuclear medicine is an expanding segment of today's medical and pharmaceutical communities. Specific radioactive isotopes are vital, with molybdenum-99 (99Mo) being the most important medical isotope. -
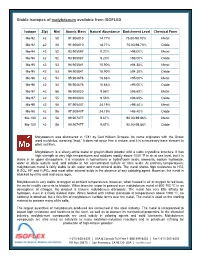
Stable Isotopes of Molybdenum Available from ISOFLEX
Stable isotopes of molybdenum available from ISOFLEX Isotope Z(p) N(n) Atomic Mass Natural Abundance Enrichment Level Chemical Form Mo-92 42 50 91.906810 14.77% 75.00-98.70% Metal Mo-92 42 50 91.906810 14.77% 75.00-98.70% Oxide Mo-94 42 52 93.905087 9.23% >98.00% Metal Mo-94 42 52 93.905087 9.23% >98.00% Oxide Mo-95 42 53 94.905841 15.90% ≥94.30% Metal Mo-95 42 53 94.905841 15.90% ≥94.30% Oxide Mo-96 42 54 95.904678 16.68% >95.00% Metal Mo-96 42 54 95.904678 16.68% >95.00% Oxide Mo-97 42 55 96.906020 9.56% ≥96.60% Metal Mo-97 42 55 96.906020 9.56% ≥96.60% Oxide Mo-98 42 56 97.905407 24.19% >98.40% Metal Mo-98 42 56 97.905407 24.19% >98.40% Oxide Mo-100 42 58 99.907477 9.67% 90.00-99.86% Metal Mo-100 42 58 99.907477 9.67% 90.00-99.86% Oxide Molybdenum was discovered in 1781 by Carl William Scheele. Its name originates with the Greek word molybdos, meaning “lead.” It does not occur free in nature, and it is a necessary trace element in plant nutrition. Molybdenum is a silvery-white metal or grayish-black powder with a cubic crystalline structure. It has high strength at very high temperatures and oxidizes rapidly above 1000 °F in air at sea level, but it is stable in an upper atmosphere. -

PSI • Scientific Report 1999 /Volume I
CH0000002 PAUL SCHERRER INSTITUT ISSN 1423-7296 March 2000 PSI • Scientific Report 1999 /Volume I Particles and Matter 3 1/28 An international collaboration of radiochemists led by the Laboratory for Radiochemistry and Environmental Chemistry of PSl and Bern University succeeded for the first time to experimentally investigate the chemical properties of bohrium (element 107) and to establish it as a member of group 7 of the Periodic Table. During a one-month long experiment, a total of only six bohrium atoms were gaschromatographically isolated in the form of volatile bohrium oxychloride molecules and identified by registering their unique decay sequence of alpha particle emissions via dubnium (element 105) to lawrencium (element 103). The short-lived bohrium atoms, decaying with a half-life of about 20 s, were produced by bombarding a very rare, highly radioactive berkelium target supplied by the US department of energy with an intense beam of neon ions at the PSl injector 1 cyclotron. PAUL SCHERRER INSTITUT ISSN 1423-7296 March 2000 \} Scientific Report 1999 Volume I Particles and Matter ed. by: J. Gobrecht, H. Gaggeler, D. Herlach, K. Junker, P.-R. Kettle, P. Kubik, A. Zehnder CH-5232 Villigen PSI Switzerland Telephone:+41 56 310 21 11 Telefax:+ 41 56 310 21 99 http://www.psi.ch TABLE OF CONTENTS Introduction .1 Laboratory for Particle Physics 3 Foreword 4 Particle Physics Theory Theory (I) 5 Theory (II) 6 Ring Accelerator Experiments Particle Properties and Decays A precise measurement of the Jt+-^7t°e+v decay rate 7 Measurement of -

Molybdenum in the Open Cluster Stars
MOLYBDENUM IN THE OPEN CLUSTER STARS T. Mishenina1, E. Shereta1, M. Pignatari2,3,6,7, G. Carraro4, T. Gorbaneva1, C. Soubiran5 1Astronomical Observatory, Odessa National University, Marazlievskaia Str., 1V, 65014, Odessa, Ukraine [email protected] 2 E.A. Milne Centre for Astrophysics, Department of Physics & Mathematics, University of Hull, HU6 7RX, United Kingdom [email protected] 3 Konkoly Observatory, Hungarian Academy of Sciences, Konkoly Thege Miklos ut 15-17, H-1121 Budapest, Hungary 4 Dipartimento di Fisica e Astronomia, Universita di Padova, I-35122, Padova, Italy [email protected] 5 Laboratoire d’astrophysique de Bordeaux, Universit´eBordeaux, CNRS, B18N, all´ee Geoffroy Saint-Hilaire, 33615 Pessac, France [email protected] 6 Joint Institute for Nuclear Astrophysics - Center for the Evolution of the Elements, USA 7 NuGrid Collaboration https://nugrid.github.io/ June 9, 2020 arXiv:2006.04629v1 [astro-ph.GA] 8 Jun 2020 Abstract Molybdenum abundances in the stars from 13 different open clusters were determined. High-resolution stellar spectra were obtained using the VLT tele- scope equipped with the UVES spectrograph on Cerro Paranal, Chile. The Mo abundances were derived in the LTE approximation from the Mo I lines at 5506 A˚ and 5533 A.˚ A comparative analysis of the behaviour of molyb- denum in the sampled stars of open clusters and Galactic disc show similar trends of decreasing Mo abundances with increasing metallicities; such a be- haviour pattern suggests a common origin of the examined populations. On the other hand, the scatter of Mo abundances in the open cluster stars is slightly greater, 0.14 dex versus 0.11 dex. -

HOT ISOSTATIC PRESSING of NANOSIZED WC-Co HARDMETALS
AT0100438 . Azcona et al. HM 6 35^ 15!h International Plansee Seminar, Eds. G. Kneringer, P. Rodhammer and H. Wildner, Plansee Holding AG, Reutte (2001), Vol. 2 HOT ISOSTATIC PRESSING OF NANOSIZED WC-Co HARDMETALS I. Azcona1'2, A. Ordonez1'2, L. Dominguez2, J.M. Sanchez1'2 and F. Castro1'2 1- CEIT - Centro de Estudios e Investigaciones Tecnicas de Guipuzcoa. Paseo Manuel de Lardizabal, 15, 20018 - San Sebastian - Spain. 2- Escuela Superior de Ingenieros de San Sebastian, Universidad de Navarra (UN). Paseo Manuel de Lardizabal, 1, 20018 - San Sebastian - Spain SUMMARY A new technique based on Hot Isostatic Pressing (HIP) has been developed to produce dense nanosized WC-Co hardmetals without the addition of grain growth inhibitors. The glass encapsulation process is the key for the effective application of isostatic pressure at temperatures well below those usually required for reaching the closed porosity state in the WC-Co system. Fully dense WC-Co samples with cobalt contents ranging from 10 to 12 wt. % have been obtained by this technique at temperatures between 1000°C and 1200°C with 150 MF'a of applied isostatic pressure for 30 minutes. The role of isostatic pressure on the activation of densification mechanisms is discussed. KEYWORDS: Hardmetals, Tungsten Carbide, Hot Isostatic Pressing, Nanocrystalline Structure. 1. INTRODUCTION Presently, the use of nanosized WC powders for producing WC-Co materials with finer microstructures represents one of the most active fields of research in the hardmetal industry. These so-called superfine grained hardmetals, with grain WC grain sizes from 0.3-0.4 \im, show not only a significant increase in hardness with respect to the finest commercial WC-Co grades (so far, with WC mean grain sizes from 0.4 to 0.5 \xm), but also higher strength and toughness (1-6). -

PRODUCTION of Mo-99/Tc-99M VIA PHOTONEUTRON REACTION USING
PRODUCTION OF Mo-99/Tc-99m VIA PHOTONEUTRON REACTION USING NATURAL MOLYBDENUM A Thesis by TALAL BADER HARAHSHEH Submitted to the Office of Graduate and Professional Studies of Texas A&M University in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Chair of Committee, Gamal Akabani Co-Chair of Committee, John W. Poston, Sr. Committee Member, Victor M. Ugaz Head of Department, Yassin A. Hassan May 2017 Major Subject: Nuclear Engineering Copyright 2017 Talal Harahsheh ABSTRACT The current shortage of the widely used radionuclide 99mTc has raised many concerns in the nuclear medicine industry. The shortage is caused by the outdated method of production using highly enriched uranium (HEU) and nuclear reactors. This production method is no longer feasible due to the restrictions on the use of HEU. Alternative methods of production must be addressed to ensure the uninterrupted supply of this key radionuclide. This research analyzes an alternative production method of 99mTc using an accelerator-generated 99Mo, which is the parent element of 99mTc. The radionuclide 99mTc has a short half-life of 6 hours. However, 99Mo, the parent nuclide has a much longer half-life of 66 hours. Usually, a medical facility receives a 99Mo/99mTc generator. Technetium-99m would be obtained from the 99Mo provided through chemical separation, which is conducted at the medical facilities. The main issues to be addressed include the production of 99Mo using the photonuclear reaction, the containment and exclusion of unwanted decay products, ensuring the integrity of the end-product 99Mo/99mTc post chemical separation, and assessing the economic feasibility of the complete strategy, including life cycle. -

Influence of Hot Isostatic Pressing On
JMEPEG ÓASM International DOI: 10.1007/s11665-014-0869-z 1059-9495/$19.00 Influence of Hot Isostatic Pressing on the Microstructure and Mechanical Properties of a Spray-Formed Al-4.5 wt.% Cu Alloy S. Devaraj, S. Sankaran, R. Kumar, and G. Appa Rao (Submitted September 6, 2013; in revised form December 4, 2013) Al-4.5 wt.% Cu alloy was spray atomized and deposited at varied spray heights ranging from 300 to 390 mm. The average grain sizes decreased from 29 to 18 lm and a concomitant increase in the hardness and the 0.2% yield strength (YS) with increase in the spray height. The respective hardness values of SF-300, SF-340, and SF-390 are 451 ± 59, 530 ± 39, and 726 ± 39 MPa and the YS are 108 ± 7, 115 ± 8, and 159 ± 10 MPa. The transmission electron micrographs revealed the morphological changes of the Al2Cu phase from irregular shaped to small plate-shaped and then subsequently to spheroidal shape due to high undercooling encountered during spray atomization with increase in spray height from 300 to 390 mm. The porosity of the spray formed deposits varied between 5 to 12%. Hot isostatic pressing of spray deposits reduced the porosity to less than 0.5% without any appreciable increase in grain size. A dislocation creep mechanism seems to be operative during the secondary processing. A comparison between as-spray formed and hot isostatically pressed deposits exemplifies improvement in mechanical properties as a result of elimination of porosity without affecting the fine grain sizes achieved during the spray-forming process. -

The Effect of Hot Isostatic Pressing on Surface Integrity, Microstructure And
applied sciences Article The Effect of Hot Isostatic Pressing on Surface Integrity, Microstructure and Strength of Hybrid Metal Injection Moulding, and Laser-Based Powder Bed Fusion Stainless-Steel Components Aldi Mehmeti 1,*, Donal Lynch 1, Pavel Penchev 1, Rafael Martinez Ramos 1, Denis Vincent 2, Johannes Maurath 3, David Ian Wimpenny 4, Khamis Essa 1 and Stefan Dimov 1,* 1 School of Mechanical Engineering, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK; [email protected] (D.L.); [email protected] (P.P.); [email protected] (R.M.R.); [email protected] (K.E.) 2 CEA, LITEN, DTNM, SA3D, LFM, Université Grenoble Alpes, F-38000 Grenoble, France; [email protected] 3 MIMplus Technologies, Turnstraße 22, 75228 Ispringen, Germany; [email protected] 4 MTC, Ansty Park, Coventry CV7 9JU, UK; [email protected] * Correspondence: [email protected] (A.M.); [email protected] (S.D.) Abstract: Hybrid manufacture of components by combining capabilities of replication and additive manufacturing processes offer a flexible and sustainable route for producing cost-effectively small batches of metal parts. At present, there are open issues related to surface integrity and performance Citation: Mehmeti, A.; Lynch, D.; of such parts, especially when utilising them in safety critical applications. The research presented Penchev, P.; Martinez Ramos, R.; in this paper investigates the ductility amplification of hybrid components produced using metal Vincent, D.; Maurath, J.; Wimpenny, injection moulding to preform and then build on them customisable sections by laser-based powder D.I.; Essa, K.; Dimov, S. -

Australian Atomic Energy Commission Research Establishment
AAEC/E644 AUSTRALIAN ATOMIC ENERGY COMMISSION RESEARCH ESTABLISHMENT LUCAS HEIGHTS RESEARCH LABORATORIES EVALUATION OF NEUTRON ACTIVATION ANALYSIS FOR THE MEASUREMENT OF ISOTOPIC ABUNDANCES IN MOLYBDENUM-98 ENRICHED MOLYBDENUM by E. L. R. HETHERINGTON MARCH 1987 ISBN 0 642 59847 9 AUSTRALIAN ATOMIC ENERGY COMMISSION RESEARCH ESTABLISHMENT LUCAS HEIGHTS RESEARCH LABORATORIES EVALUATION OF NEUTRON ACTIVATION ANALYSIS FOR THE MEASUREMENT OF ISOTOPIC ABUNDANCES IN MOLYBDENUM-98 ENRICHED MOLYBDENUM by E.L.R. HETHERINGTON ABSTRACT Molybdenum enriched to approximately 50 per cent 98Mo is required for the development of a portable 99mTc generator based on the zirconium molybdate gel system developed by the AAEC. It has been proposed that existing AAEC facilities be used to enrich molybdenum, and neutron activation analysis has been evaluated as a means of monitoring the enrichment process. 98 Simulated enriched targets of Mo03 with Mo abundances ranging from 30 to 90 per cent were prepared by 98 diluting 96.87 per cent enriched MoO3 with the natural trioxide. These targets were each irradiated 12 2 1 simultaneously with a reference natural Mo03 target in a thermal neutron flux of 7.5 x 10 neutrons cm~ s~ where the reactions 92Mo(n/y) 93Mo, 98Mo(n,Y) 99Mo and 100Mo(n,Y)101Mo-101Tc occur. The isotopic abundances of 98Mo and 100Mo were determined with an accuracy ± 2 per cent by comparing the yields of 99Mo-99mTc and 101Mo-101Tc, respectively, in the enriched and natural targets. The activation yield of 93Mo was too low for the determination of the 92Mo abundances. National Library of Australia card number and ISBN 0 642 59847 9 The following descriptors have been selected from the INIS Thesaurus to describe the subject content of this report for Information retrieval purposes. -

Potential Production of Molybdenum-99
DEVELOPMENT OF A 37-ELEMENT FUEL BUNDLE FOR THE PRODUCTION OF MOLYBDENUM-99 IN CANDU POWER REACTORS By Jawad Haroon A Thesis Submitted in Partial Fulfillment of the Requirements for the Degree of Master of Applied Science In The Faculty of Energy Systems and Nuclear Science Nuclear Engineering University of Ontario Institute of Technology August 2014 © Jawad Haroon, 2014 Abstract In this study, the potential use of CANDU power reactors for the production of Mo-99 is assessed. Five different modifications of a 37-element fuel bundle that could be used for the production of Mo-99 in existing CANDU type power reactors are explored. Since Mo-99 is generated by the fission of U-235 with a fission yield of 6.1%, the proposed designs use enriched uranium in the Mo-99 producing fuel pins. The challenge lies in designing a fuel that is able to produce significant quantities of Mo-99 while having similar neutronics and thermalhydraulic characteristics to the standard CANDU fuel. The proposed designs, when irradiated in the peak power channel of a CANDU core, are shown to produce significant quantities of Mo-99 while maintaining the necessary reactivity and power rating limits. The total Mo-99 production activities are found to be approximately 2335, 2297, 2336, 4131 and 4268 six-day Curies per bundle for Designs 1 to 5, respectively. The yield corresponds to approximately 19% (for Designs 1, 2 and 3) and 34% (for Designs 4 and 5) of the world weekly demand for Mo-99. A production cycle of 6 bundles per week (for Designs 1, 2 and 3), or 3 bundles per week (for Designs 4 and 5) can meet the global demand of Mo-99 medical isotopes. -

INFORMATION from ISOTOPE RATIOS O. Manuel(1)
Proc. of 2002 SOHO 12 /GONG + 2002, Local and Global Helioseismology: The Present and Future, Big Bear Lake, CA USA (Ed: Huguette Lacoste, ESA SP-517, Feb 2003) pp. 345-348 COMPOSITION OF THE SOLAR INTERIOR: INFORMATION FROM ISOTOPE RATIOS O. Manuel(1) and Stig Friberg(2) 1University of Missouri, Nuclear Chemistry, Rolla, MO 65409 U.S.A. tel: +1 573-341-4420 / fax: +1 573-341-6033 / e-mail: [email protected] 2Clarkson University, Physical Chemistry, 641 Spring Hill Estate, Eminence, KY 13699-5814 U.S.A. ABSTRACT confirmed in diverse types of meteorites [10,11]. The amount of Xe-2 in the early solar system was Measurements are reviewed showing that the interior sufficient to shift the composition of bulk xenon in of the Sun, the inner planets, and ordinary meteorites meteorites (Point AVCC in Fig. 1) away from Xe-1. consist mostly of the same elements: Iron, oxygen, nickel, silicon, magnesium, sulfur and calcium [1]. These results do not support the standard solar model. INTRODUCTION Reynolds found the decay product of extinct 129I and an unusual abundance pattern of the other xenon isotopes in meteorites in 1960 [2,3]. Since 129I exceeded that expected if the solar system formed from an interstellar cloud, Fowler et al. [4] suggested that D, Li, Be, B and extinct 129I and 107Pd might have been produced locally in the early solar system. Figure 2: The link of primordial helium with xenon In 1965 the decay product of extinct 244Pu [5], a isotopes in the Allende meteorite [9]. nuclide that could only be made by rapid neutron- capture in a supernova (SN), was found in meteorites.