Phosphotriesterase: an Enzyme in Search of Its Natural Substrate
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Classification of Esterases: an Important Gene Family Involved in Insecticide Resistance - a Review
Mem Inst Oswaldo Cruz, Rio de Janeiro, Vol. 107(4): 437-449, June 2012 437 The classification of esterases: an important gene family involved in insecticide resistance - A Review Isabela Reis Montella1,2, Renata Schama1,2,3/+, Denise Valle1,2,3 1Laboratório de Fisiologia e Controle de Artrópodes Vetores, Instituto Oswaldo Cruz-Fiocruz, Av. Brasil 4365, 21040-900 Rio de Janeiro, RJ, Brasil 2Instituto de Biologia do Exército, Rio de Janeiro, RJ, Brasil 3Instituto Nacional de Ciência e Tecnologia em Entomologia Molecular, Rio de Janeiro, RJ, Brasil The use of chemical insecticides continues to play a major role in the control of disease vector populations, which is leading to the global dissemination of insecticide resistance. A greater capacity to detoxify insecticides, due to an increase in the expression or activity of three major enzyme families, also known as metabolic resistance, is one major resistance mechanisms. The esterase family of enzymes hydrolyse ester bonds, which are present in a wide range of insecticides; therefore, these enzymes may be involved in resistance to the main chemicals employed in control programs. Historically, insecticide resistance has driven research on insect esterases and schemes for their classification. Currently, several different nomenclatures are used to describe the esterases of distinct species and a universal standard classification does not exist. The esterase gene family appears to be rapidly evolving and each insect species has a unique complement of detoxification genes with only a few orthologues across species. The examples listed in this review cover different aspects of their biochemical nature. However, they do not appear to contribute to reliably distinguish among the different resistance mechanisms. -

Supporting Information
Supporting Information Figure S1. The functionality of the tagged Arp6 and Swr1 was confirmed by monitoring cell growth and sensitivity to hydeoxyurea (HU). Five-fold serial dilutions of each strain were plated on YPD with or without 50 mM HU and incubated at 30°C or 37°C for 3 days. Figure S2. Localization of Arp6 and Swr1 on chromosome 3. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 3L, Tel 3R, CEN3, and the RP gene are shown under the panels. Figure S3. Localization of Arp6 and Swr1 on chromosome 4. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) in the whole chromosome region are compared. The position of Tel 4L, Tel 4R, CEN4, SWR1, and RP genes are shown under the panels. Figure S4. Localization of Arp6 and Swr1 on the region including the SWR1 gene of chromosome 4. The binding of Arp6- FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position and orientation of the SWR1 gene is shown. Figure S5. Localization of Arp6 and Swr1 on chromosome 5. The binding of Arp6-FLAG (top), Swr1-FLAG (middle), and Arp6-FLAG in swr1 cells (bottom) are compared. The position of Tel 5L, Tel 5R, CEN5, and the RP genes are shown under the panels. Figure S6. Preferential localization of Arp6 and Swr1 in the 5′ end of genes. Vertical bars represent the binding ratio of proteins in each locus. -

Reduction of Pectinesterase Activity in a Commercial Enzyme Preparation
Journal of the Science of Food and Agriculture J Sci Food Agric 85:1613–1621 (2005) DOI: 10.1002/jsfa.2154 Reduction of pectinesterase activity in a commercial enzyme preparation by pulsed electric fields: comparison of inactivation kinetic models Joaquın´ Giner, Pascal Grouberman, Vicente Gimeno and Olga Martın´ ∗ Department of Food Technology, University of Lleida, CeRTA-UTPV, ETSEA, Avda Alcalde Rovira Roure 191, 25198-Lleida, Spain Abstract: The inactivation of pectinesterase (PE) in a commercial enzyme preparation (CEP) under high intensity pulsed electric fields (HIPEF) was studied. After desalting and water dilution of the raw CEP, samples were exposed to exponentially decay waveform pulses for up to 463 µs at electric field intensities ranging from 19 to 38 kV cm−1. Pulses were applied in monopolar mode. Experimental data were fitted to a first-order kinetic model as well as to models based on Fermi, Hulsheger¨ or Weibull equations to describe PE inactivation kinetics. Characteristic parameters for each model were calculated. Relationships between some of the parameters and process variables were obtained. The Weibull model yielded the best accuracy factor. The relationship between residual PE and input of electrical energy density was found to be that of exponential decay. 2005 Society of Chemical Industry Keywords: pulsed electric fields; kinetics; pectinesterase; model; inactivation INTRODUCTION It has become customary to use CEPs in fruit and Pectinesterase (PE; EC 3.1.1.11) is a pectic enzyme vegetable juice technology. Depending -

Download Product Insert (PDF)
PRODUCT INFORMATION Lysophospholipase D Polyclonal Antibody Item No. 10005375 Overview and Properties Contents: This vial contains 500 µl of peptide affinity-purified antibody. Synonyms: Autotaxin, ENPP2, lysoPLD Immunogen: Peptide from the C-terminal region of rat LysoPLD Species Reactivity: (+) Human, mouse, and rat; other species not tested Form: Liquid Storage: -20°C (as supplied) Stability: ≥1 year Storage Buffer: TBS, pH 7.4, with 50% glycerol, 0.1%l BSA, and 0.02% sodium azide Host: Rabbit Applications: Immunocytochemistry (ICC), Immunohistochemistry (IHC), and Western blot (WB); the recommended starting dilution for ICC is 1:500, 1:80 for IHC, and 1:200 for WB. Other applications were not tested, therefore optimal working concentration/dilution should be determined empirically. Images 1 · · · · · · · 104 kDa · · · · · · · 60 kDa Lane 1: Human cerebella supernatant (40 µg) Immunohistochemistry analysis of formalin-fixed, paraffin-embedded (FFPE) human cerebellum ǎssue aer heat-induced anǎgen retrieval in pH 6.0 citrate buffer. Aer incubaǎon with Lysophospholipase D Polyclonal Anǎbody (Item No. 10005375) at a 1:80 diluǎon, slides were incubated with bioǎnylated secondary anǎbody, followed by alkaline phosphatase-strepavidin and chromogen (DAB). WARNING CAYMAN CHEMICAL THIS PRODUCT IS FOR RESEARCH ONLY - NOT FOR HUMAN OR VETERINARY DIAGNOSTIC OR THERAPEUTIC USE. 1180 EAST ELLSWORTH RD SAFETY DATA ANN ARBOR, MI 48108 · USA This material should be considered hazardous until further information becomes available. Do not ingest, inhale, get in eyes, on skin, or on clothing. Wash thoroughly after handling. Before use, the user must review the complete Safety Data Sheet, which has been sent via email to your institution. PHONE: [800] 364-9897 WARRANTY AND LIMITATION OF REMEDY [734] 971-3335 Buyer agrees to purchase the material subject to Cayman’s Terms and Conditions. -

Diagnostic Value of Serum Enzymes-A Review on Laboratory Investigations
Review Article ISSN 2250-0480 VOL 5/ ISSUE 4/OCT 2015 DIAGNOSTIC VALUE OF SERUM ENZYMES-A REVIEW ON LABORATORY INVESTIGATIONS. 1VIDYA SAGAR, M.SC., 2DR. VANDANA BERRY, MD AND DR.ROHIT J. CHAUDHARY, MD 1Vice Principal, Institute of Allied Health Sciences, Christian Medical College, Ludhiana 2Professor & Ex-Head of Microbiology Christian Medical College, Ludhiana 3Assistant Professor Department of Biochemistry Christian Medical College, Ludhiana ABSTRACT Enzymes are produced intracellularly, and released into the plasma and body fluids, where their activities can be measured by their abilities to accelerate the particular chemical reactions they catalyze. But different serum enzymes are raised when different tissues are damaged. So serum enzyme determination can be used both to detect cellular damage and to suggest its location in situ. Some of the biochemical markers such as alanine aminotransferase, aspartate aminotransferase, alkaline phasphatase, gamma glutamyl transferase, nucleotidase, ceruloplasmin, alpha fetoprotein, amylase, lipase, creatine phosphokinase and lactate dehydrogenase are mentioned to evaluate diseases of liver, pancreas, skeletal muscle, bone, etc. Such enzyme test may assist the physician in diagnosis and treatment. KEYWORDS: Liver Function tests, Serum Amylase, Lipase, CPK and LDH. INTRODUCTION mitochondrial AST is seen in extensive tissue necrosis during myocardial infarction and also in chronic Liver diseases like liver tissue degeneration DIAGNOSTIC SERUM ENZYME and necrosis². But lesser amounts are found in Enzymes are very helpful in the diagnosis of brain, pancreas and lung. Although GPT is plentiful cardiac, hepatic, pancreatic, muscular, skeltal and in the liver and occurs only in the small amount in malignant disorders. Serum for all enzyme tests the other tissues. -

744 Hydrolysis of Chiral Organophosphorus Compounds By
[Frontiers in Bioscience, Landmark, 26, 744-770, Jan 1, 2021] Hydrolysis of chiral organophosphorus compounds by phosphotriesterases and mammalian paraoxonase-1 Antonio Monroy-Noyola1, Damianys Almenares-Lopez2, Eugenio Vilanova Gisbert3 1Laboratorio de Neuroproteccion, Facultad de Farmacia, Universidad Autonoma del Estado de Morelos, Morelos, Mexico, 2Division de Ciencias Basicas e Ingenierias, Universidad Popular de la Chontalpa, H. Cardenas, Tabasco, Mexico, 3Instituto de Bioingenieria, Universidad Miguel Hernandez, Elche, Alicante, Spain TABLE OF CONTENTS 1. Abstract 2. Introduction 2.1. Organophosphorus compounds (OPs) and their toxicity 2.2. Metabolism and treatment of OP intoxication 2.3. Chiral OPs 3. Stereoselective hydrolysis 3.1. Stereoselective hydrolysis determines the toxicity of chiral compounds 3.2. Hydrolysis of nerve agents by PTEs 3.2.1. Hydrolysis of V-type agents 3.3. PON1, a protein restricted in its ability to hydrolyze chiral OPs 3.4. Toxicity and stereoselective hydrolysis of OPs in animal tissues 3.4.1. The calcium-dependent stereoselective activity of OPs associated with PON1 3.4.2. Stereoselective hydrolysis commercial OPs pesticides by alloforms of PON1 Q192R 3.4.3. PON1, an enzyme that stereoselectively hydrolyzes OP nerve agents 3.4.4. PON1 recombinants and stereoselective hydrolysis of OP nerve agents 3.5. The activity of PTEs in birds 4. Conclusions 5. Acknowledgments 6. References 1. ABSTRACT Some organophosphorus compounds interaction of the racemic OPs with these B- (OPs), which are used in the manufacturing of esterases (AChE and NTE) and such interactions insecticides and nerve agents, are racemic mixtures have been studied in vivo, ex vivo and in vitro, using with at least one chiral center with a phosphorus stereoselective hydrolysis by A-esterases or atom. -

Enzymatic Degradation of Organophosphorus Pesticides and Nerve Agents by EC: 3.1.8.2
catalysts Review Enzymatic Degradation of Organophosphorus Pesticides and Nerve Agents by EC: 3.1.8.2 Marek Matula 1, Tomas Kucera 1 , Ondrej Soukup 1,2 and Jaroslav Pejchal 1,* 1 Department of Toxicology and Military Pharmacy, Faculty of Military Health Sciences, University of Defence, Trebesska 1575, 500 01 Hradec Kralove, Czech Republic; [email protected] (M.M.); [email protected] (T.K.); [email protected] (O.S.) 2 Biomedical Research Center, University Hospital Hradec Kralove, Sokolovska 581, 500 05 Hradec Kralove, Czech Republic * Correspondence: [email protected] Received: 26 October 2020; Accepted: 20 November 2020; Published: 24 November 2020 Abstract: The organophosphorus substances, including pesticides and nerve agents (NAs), represent highly toxic compounds. Standard decontamination procedures place a heavy burden on the environment. Given their continued utilization or existence, considerable efforts are being made to develop environmentally friendly methods of decontamination and medical countermeasures against their intoxication. Enzymes can offer both environmental and medical applications. One of the most promising enzymes cleaving organophosphorus compounds is the enzyme with enzyme commission number (EC): 3.1.8.2, called diisopropyl fluorophosphatase (DFPase) or organophosphorus acid anhydrolase from Loligo Vulgaris or Alteromonas sp. JD6.5, respectively. Structure, mechanisms of action and substrate profiles are described for both enzymes. Wild-type (WT) enzymes have a catalytic activity against organophosphorus compounds, including G-type nerve agents. Their stereochemical preference aims their activity towards less toxic enantiomers of the chiral phosphorus center found in most chemical warfare agents. Site-direct mutagenesis has systematically improved the active site of the enzyme. These efforts have resulted in the improvement of catalytic activity and have led to the identification of variants that are more effective at detoxifying both G-type and V-type nerve agents. -
What Is Alkaline Phosphatase? What Are the Symptoms of Low ALP?
What is the treatment for HPP? In 2015, asfotase alfa (Strensiq™) was approved for use in the US, the European Union, and Canada for pediatric-onset HPP, and in Japan for HPP with onset at any age. The medication is an injection given multiple times per week underneath the skin (subcutaneous). It is a recombinant (factory-made) form of ALP that has a bone-targeting component. When patients take asfotase alfa, ALP levels measured in the blood are quite high (often in the multiple thousands). Therefore, measuring ALP levels is typically not helpful once a patient is on therapy. What is Alkaline Phosphatase? What are the symptoms of low ALP? For more information, please contact Patients with hypophosphatasia (HPP) have Low ALP can lead to multiple symptoms of HPP, the Soft Bones Foundation. a low blood alkaline phosphatase (ALP) level. including poor bone mineralization and rickets as well (866) 827-9937 – Toll Free as early tooth loss (prior to age 5 years). It is also (973) 453-3093 – Direct Line thought that PPi may build up in muscle tissue 121 Hawkins Place, #267 ALP is an enzyme (a protein that breaks down and may be responsible for the pain and muscle Boonton, New Jersey 07005 chemicals). Most health providers are aware that weakness that some patients with HPP experience. www.SoftBones.org high ALP levels may indicate medical problems, such as liver disease or other bone diseases besides HPP, but many providers may not recognize that low ALP levels can indicate HPP. Low ALP is important What questions should patients ask Written by because the inability to break down chemicals can their doctors about ALP? Jill Simmons, MD lead to elevated levels of these chemicals, which Associate Professor of Pediatrics, Ian Burr Division can cause multiple problems. -

Effect of Alkaline Phosphatase on the Function of Purified Reverse Transcriptase in Reconstructed Reverse Transcription (116 Pp) Director: Kenneth F
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 1984 Effect of alkaline phosphatase on the function of purified er verse transcriptase in reconstructed reverse transcription Michael J. Wolkowicz The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Wolkowicz, Michael J., "Effect of alkaline phosphatase on the function of purified er verse transcriptase in reconstructed reverse transcription" (1984). Graduate Student Theses, Dissertations, & Professional Papers. 7480. https://scholarworks.umt.edu/etd/7480 This Thesis is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. COPYRIGHT ACT OF 1976 This is an unpublished manuscript in which copyright sub s i s t s . Any further reprinting of its contents must be approved BY THE a u t h o r . Mansfield Library University of Montana Date ; 1 G 8 5 EFFECT OF ALKALINE PHOSPHATASE ON THE FUNCTION OF PURIFIED REVERSE TRANSCRIPTASE IN RECONSTRUCTED REVERSE TRANSCRIPTION By Michael J. Wolkowicz B. A. American International College, 1976 B. S. University of Montana, 1980 Presented in partial fulfillment of the requirements for the degree of Masters of Science UNIVERSITY OF MONTANA 1984 Approved by: iP- airman. Board of Examiners D^n, Graduater'school Date UMI Number: EP38281 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. -
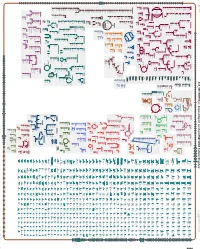
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

The Characterization of Oligonucleotides and Nucleic Acids Using Ribonuclease H and Mass Spectrometry
Louisiana State University LSU Digital Commons LSU Historical Dissertations and Theses Graduate School 1999 The hC aracterization of Oligonucleotides and Nucleic Acids Using Ribonuclease H and Mass Spectrometry. Lenore Marie Polo Louisiana State University and Agricultural & Mechanical College Follow this and additional works at: https://digitalcommons.lsu.edu/gradschool_disstheses Recommended Citation Polo, Lenore Marie, "The hC aracterization of Oligonucleotides and Nucleic Acids Using Ribonuclease H and Mass Spectrometry." (1999). LSU Historical Dissertations and Theses. 6923. https://digitalcommons.lsu.edu/gradschool_disstheses/6923 This Dissertation is brought to you for free and open access by the Graduate School at LSU Digital Commons. It has been accepted for inclusion in LSU Historical Dissertations and Theses by an authorized administrator of LSU Digital Commons. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter free, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand corner and continuing from left to right in equal sections with small overlaps. -

Reconsideration of the Catalytic Center and Mechanism of Mammalian
Proc. Natl. Acad. Sci. USA Vol. 92, pp. 7187-7191, August 1995 Genetics Reconsideration of the catalytic center and mechanism of mammalian paraoxonase/arylesterase (organophosphatase/A-esterase/site-directed mutagenesis/high density lipoprotein/phosphotriesterase) ROBERT C. SORENSON*, SERGIO L. PRIMO-PARMOt, CHUNG-LIANG Kuot, STEVE ADKINS0§, OKSANA LOCKRIDGEI, AND BERT N. LA DU*tll *Department of Pharmacology, University of Michigan Medical School, Ann Arbor, MI 48109-0632; tDepartment of Anesthesiology, Research Division, 4038 Kresge Research II, University of Michigan Medical School, Ann Arbor, MI 48109-0572; tDepartment of Environmental and Industrial Health, School of Public Health, University of Michigan, Ann Arbor, MI 48109-2029; §Mental Health Research Institute and Department of Human Genetics, University of Michigan Medical School, Ann Arbor, MI 48109-0720; and lEppley Institute, University of Nebraska Medical Center, Omaha, NB 68198-6805 Communicated by James V Neel, University ofMichigan Medical School, Ann Arbor, MI, May 2, 1995 ABSTRACT For three decades, mammalian paraoxonase basis for the human polymorphism was the presence of argi- (A-esterase, aromatic esterase, arylesterase; PON, EC 3.1.8.1) nine (high PON activity, B-type allozyme) or glutamine (low has been thought to be a cysteine esterase demonstrating PON activity, A-type allozyme) at position 191 (11-13). structural and mechanistic homologies with the serine ester- Augustinsson (14, 15) suggested that PON, the carboxyes- ases (cholinesterases and carboxyesterases). Human, mouse, terases, and the cholinesterases were products of divergent and rabbit PONs each contain only three cysteine residues, evolution from an ancestral arylesterase but that PON utilized and their positions within PON have been conserved.