Profiling the Toxcast Library with a Pluripotent Human (H9) Stem Cell Line-Based Biomarker Assay for Developmental Toxici- Ty[AQ2][AQ3]
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
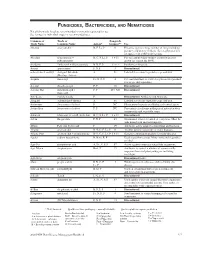
Fungicides, Bactericides, and Nematicides Not All Chemicals Listed Are Recommended Or Currently Registered for Use
FUNGICIDES, BACTERICIDES, AND NEMATICIDES Not all chemicals listed are recommended or currently registered for use. See listings for individual crops for recommended uses. Common or Trade or Fungicide Trade Name Common Name Action* Group #** Use Abound azoxystrobin B, F, Ls, P 11 Effective against a large number of fungi including powdery and downy mildews. Severe phytotoxicity on apples with a McIntosh heritage. Absolute tebuconazole + B, C, F, Ls, P 3 + 11 For rust and powdery mildew control in grasses trifloxystrobin grown for seed in the PNW. Academy fludioxonil + difenoconazole B-N, F, P 12 + 3 Postharvest fungicide. Accrue spiroxamine F, N, P 5 Discontinued. acibenzolar-S-methyl Actigard, Blockade A P1 Labeled for certain vegetable crops and fruit. (Heritage Action) Acquire metalaxyl Fs, N, P, S 4 For seed treatment to control ooymcetes in specified row crops and vegetables. Acrobat dimethomorph F, P 40 Discontinued. Acrobat MZ dimethomorph + F, P 40 + M3 Discontinued. mancozeb Acti-dione cycloheximide F Discontinued. Antibiotic and fungicide. Actigard acibenzolar-S-methyl A P1 Labeled for certain vegetable crops and fruit. Actinovate Streptomyces lydicus F NC Filamentous bacteria as a Biological control agent. Actino-Iron Streptomyces lydicus F, P NC For control of soilborne pathogens of indoor/outdoor ornamentals and vegetable crops. Adament tebuconazole + trifloxystrobin B, C, F, Ls, P 3 + 11 Discontinued. Adorn fluopicolide F, N, P 43 Ornamental label for control of oomycetes. Must be tank-mixed with another fungicide. Affirm Polyoxin D zinc salt F 19 Antibiotic active against certain fungi and bacteria. Aframe azoxystrobin B-N, C, F, Ls, P 11 Another generic fungicide for many diseases. -

Casein Kinase 1 Isoforms in Degenerative Disorders
CASEIN KINASE 1 ISOFORMS IN DEGENERATIVE DISORDERS DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Theresa Joseph Kannanayakal, M.Sc., M.S. * * * * * The Ohio State University 2004 Dissertation Committee: Approved by Professor Jeff A. Kuret, Adviser Professor John D. Oberdick Professor Dale D. Vandre Adviser Professor Mike X. Zhu Biophysics Graduate Program ABSTRACT Casein Kinase 1 (CK1) enzyme is one of the largest family of Serine/Threonine protein kinases. CK1 has a wide distribution spanning many eukaryotic families. In cells, its kinase activity has been found in various sub-cellular compartments enabling it to phosphorylate many proteins involved in cellular maintenance and disease pathogenesis. Tau is one such substrate whose hyperphosphorylation results in degeneration of neurons in Alzheimer’s disease (AD). AD is a slow neuroprogessive disorder histopathologically characterized by Granulovacuolar degeneration bodies (GVBs) and intraneuronal accumulation of tau in Neurofibrillary Tangles (NFTs). The level of CK1 isoforms, CK1α, CK1δ and CK1ε has been shown to be elevated in AD. Previous studies of the correlation of CK1δ with lesions had demonstrated its importance in tau hyperphosphorylation. Hence we investigated distribution of CK1α and CK1ε with the lesions to understand if they would play role in tau hyperphosphorylation similar to CK1δ. The kinase results were also compared with lesion correlation studies of peptidyl cis/trans prolyl isomerase (Pin1) and caspase-3. Our results showed that among the enzymes investigated, CK1 isoforms have the greatest extent of colocalization with the lesions. We have also investigated the distribution of CK1α with different stages of NFTs that follow AD progression. -

Molecular Cloning of a Candidate Tumor Suppressor Gene, DLC1, from Chromosome 3P21.31
[CANCER RESEARCH 59, 1966–1972, April 15, 1999] Molecular Cloning of a Candidate Tumor Suppressor Gene, DLC1, from Chromosome 3p21.31 Yataro Daigo, Tadashi Nishiwaki, Teru Kawasoe, Mayumi Tamari, Eiju Tsuchiya, and Yusuke Nakamura2 Laboratory of Molecular Medicine, Human Genome Center, Institute of Medical Science, The University of Tokyo, Tokyo 108, Japan [Y. D., T. N., T. K., M. T., Y. N.], and Department of Pathology, Saitama Cancer Center Research Institute, Saitama, Japan [E. T.] ABSTRACT MATERIALS AND METHODS The short arm of chromosome 3 is thought to contain multiple tumor Cell Lines and Primary Tumor Samples. Fourteen human esophageal suppressor genes, because one copy of this chromosomal arm frequently is carcinoma cell lines [TE series: gifts from Dr. Tetsuro Nishihira, Tohoku missing in carcinomas that have arisen in a variety of tissues. We have University (Miyagi); Ref. 12], six lung cancer cell lines [LC319, a gift from isolated a novel gene encoding a 1755-amino acid polypeptide, through Dr. Takashi Takahashi, Aichi Cancer Center (Aichi); A549, NCI-H23, -H226, large-scale sequencing of genomic DNA at 3p21.3. Mutational analysis of -H460, -H522, gifts from Dr. Takao Yamori, Cancer Institute (Tokyo)], and this gene by reverse transcription-PCR revealed the lack of functional two renal cancer cell lines (RXF631L and ACHN, gifts from Dr. Takao transcripts and an increase of nonfunctional RNA transcripts in a signif- Yamori) were grown in monolayers in RPMI 1640 supplemented with 5–10% icant proportion (33%) of cancer cell lines and primary cancers (4 of 14 fetal bovine serum. esophageal cancer cell lines, 2 of 2 renal cancer cell lines, 11 of 30 primary Tumors and corresponding normal tissue samples were obtained from a total non-small cell lung cancers, and 3 of 10 primary squamous cell carcino- of 48 patients with NSCLCs and 10 patients with primary esophageal squa- mas of the esophagus). -

CSNK1D Monoclonal Antibody (M09), Clone 4H8
CSNK1D monoclonal antibody (M09), clone 4H8 Catalog # : H00001453-M09 規格 : [ 100 ug ] List All Specification Application Image Product Mouse monoclonal antibody raised against a partial recombinant Western Blot (Recombinant protein) Description: CSNK1D. Immunofluorescence Immunogen: CSNK1D (AAH03558, 301 a.a. ~ 415 a.a) partial recombinant protein with GST tag. MW of the GST tag alone is 26 KDa. Sequence: ADDAERERRDREERLRHSRNPATRGLPSTASGRLRGTQEVAPPTPLTP TSHTANTSPRPVSGMERERKVSMRLHRGAPVNISSSDLTGRQDTSRMS TSQIPGRVASSGLQSVVHR enlarge Host: Mouse Immunohistochemistry Reactivity: Human (Formalin/PFA-fixed paraffin- embedded sections) Isotype: IgG2a Kappa Quality Control Antibody Reactive Against Recombinant Protein. Testing: enlarge Sandwich ELISA (Recombinant protein) enlarge Western Blot detection against Immunogen (38.28 KDa) . ELISA Storage Buffer: In 1x PBS, pH 7.4 In situ Proximity Ligation Assay Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. (Cell) Instruction: MSDS: Download Interspecies Rat (99) Antigen enlarge Sequence: Datasheet: Download Applications Western Blot (Recombinant protein) Protocol Download Immunofluorescence Page 1 of 4 2021/6/19 enlarge this image Immunofluorescence of monoclonal antibody to CSNK1D on HeLa cell . [antibody concentration 10 ug/ml] Immunohistochemistry (Formalin/PFA-fixed paraffin-embedded sections) enlarge this image Immunoperoxidase of monoclonal antibody to CSNK1D on formalin-fixed paraffin- embedded human placenta. [antibody concentration 3 ug/ml] Protocol Download Sandwich ELISA (Recombinant protein) Detection limit for recombinant GST tagged CSNK1D is 0.3 ng/ml as a capture antibody. Protocol Download ELISA In situ Proximity Ligation Assay (Cell) Proximity Ligation Analysis of protein-protein interactions between TP53 and CSNK1D. HeLa cells were stained with anti-TP53 rabbit purified polyclonal 1:1200 and anti-CSNK1D mouse monoclonal antibody 1:50. -

Gene Expression Barcode Values Reveal a Potential Link Between Parkinson's Disease and Gastric Cancer
University of Kentucky UKnowledge Internal Medicine Faculty Publications Internal Medicine 2-16-2021 Gene Expression Barcode Values Reveal a Potential Link between Parkinson's Disease and Gastric Cancer Suyan Tian First Hospital of Jilin University, China Shishun Zhao Jilin University, China Mingbo Tang First Hospital of Jilin University, China Chi Wang University of Kentucky, [email protected] Follow this and additional works at: https://uknowledge.uky.edu/internalmedicine_facpub Part of the Geriatrics Commons, Internal Medicine Commons, and the Oncology Commons Right click to open a feedback form in a new tab to let us know how this document benefits ou.y Repository Citation Tian, Suyan; Zhao, Shishun; Tang, Mingbo; and Wang, Chi, "Gene Expression Barcode Values Reveal a Potential Link between Parkinson's Disease and Gastric Cancer" (2021). Internal Medicine Faculty Publications. 231. https://uknowledge.uky.edu/internalmedicine_facpub/231 This Article is brought to you for free and open access by the Internal Medicine at UKnowledge. It has been accepted for inclusion in Internal Medicine Faculty Publications by an authorized administrator of UKnowledge. For more information, please contact [email protected]. Gene Expression Barcode Values Reveal a Potential Link between Parkinson's Disease and Gastric Cancer Digital Object Identifier (DOI) https://doi.org/10.18632/aging.202623 Notes/Citation Information Published by Aging, v. 13. © 2021 Tian et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. This article is available at UKnowledge: https://uknowledge.uky.edu/internalmedicine_facpub/231 www.aging-us.com AGING 2021, Vol. -
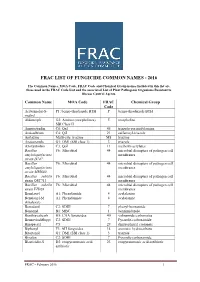
Frac List of Fungicide Common Names - 2016
FRAC LIST OF FUNGICIDE COMMON NAMES - 2016 The Common Names, MOA Code, FRAC Code and Chemical Group names included in this list are those used in the FRAC Code List and the associated List of Plant Pathogenic Organisms Resistant to Disease Control Agents. Common Name MOA Code FRAC Chemical Group Code Acibenzolar-S- P1: benzo-thiadiazole BTH P benzo-thiadiazole BTH methyl Aldimorph G2: Amines (morpholines) 5 morpholine SBI Class II Ametoctradin C8: QxI 45 triazolo-pyrimidylamine Amisulbrom C4: QiI 21 sulfamoyl-triazole Anilazine Multi-site: triazine M8 triazine Azaconazole G1: DMI (SBI class 1) 3 triazole Azoxystrobin C3: QoI 11 methoxy-acrylates Bacillus F6: Microbial 44 microbial disrupters of pathogen cell amyloliquefaciens membranes strain D747 Bacillus F6: Microbial 44 microbial disrupters of pathogen cell amyloliquefaciens membranes strain MBI600 Bacillus subtilis F6: Microbial 44 microbial disrupters of pathogen cell strain QST713 membranes Bacillus subtilis F6: Microbial 44 microbial disrupters of pathogen cell strain FZB24 membranes Benalaxyl A1: Phenylamide 4 acylalanine Benalaxyl-M A1: Phenylamide 4 acylalanine (kiralaxyl) Benodanil C2: SDHI 7 phenyl-benzamide Benomyl B1: MBC 1 benzimidazole Benthiavalicarb H5: CAA fungicides 40 valinamide carbamates Benzovindiflupyr C2: SDHI 7 Pyrazole-carboxamide Binapacryl C5 29 dinitrophenyl crotonate Biphenyl F3: AH fungicides 14 aromatic hydrocarbons Bitertanol G1: DMI (SBI class 1) 3 triazole Bixafen C2: SDHI 7 Pyrazole-carboxamide Blasticidin-S D2: enopyranuronic acid 23 enopyranuronic -

Association of Genetic Variation in Genes Implicated in the B-Catenin Destruction Complex with Risk of Breast Cancer
2101 Association of Genetic Variation in Genes Implicated in the B-Catenin Destruction Complex with Risk of Breast Cancer Xianshu Wang,1 Ellen L. Goode,2 Zachary S. Fredericksen,2 Robert A. Vierkant,2 V. Shane Pankratz,2 Wen Liu-Mares,2 David N. Rider,2 Celine M. Vachon,2 James R. Cerhan,2 Janet E. Olson,2 and Fergus J. Couch1 Departments of 1Laboratory Medicine and Pathology and 2Health Sciences Research, Mayo Clinic, Rochester, Minnesota Abstract B P Aberrant Wnt/ -catenin signaling leading to nuclear 95% confidence intervals, 1.05-1.43; trend = 0.01). In accumulation of the oncogene product B-catenin is ob- addition, five SNPs in AXIN2 were associated with in- P served in a wide spectrum of human malignancies. creased risk of breast cancer ( trend < 0.05). Haplotype- The destruction complex in the Wnt/B-catenin pathway based tests identified significant associations between is critical for regulating the level of B-catenin in the specific haplotypes in APC and AXIN2 (P V 0.03) and cytoplasm and in the nucleus. Here, we report a com- breast cancer risk. Further characterization of the APC prehensive study of the contribution of genetic varia- and AXIN2 variants suggested that AXIN2 rs4791171 tion in six genes encoding the B-catenin destruction was significantly associated with risk in premeno- APC, AXIN1, AXIN2, CSNK1D, CSNK1E P complex ( , and pausal ( trend = 0.0002) but not in postmenopausal GSK3B) to breast cancer using a Mayo Clinic Breast women. The combination of our findings and numer- Cancer Case-Control Study. A total of 79 candidate ous genetic and functional studies showing that APC functional and tagging single nucleotide polymor- and AXIN2 perform crucial tumor suppressor func- phisms (SNP) were genotyped in 798 invasive cases tions suggest that further investigation of the contri- and 843 unaffected controls. -

Casein Kinase 1D Encodes a Novel Drug Target in Hedgehog—GLI-Driven Cancers and Tumor-Initiating Cells Resistant to SMO Inhibition
cancers Article Casein Kinase 1D Encodes a Novel Drug Target in Hedgehog—GLI-Driven Cancers and Tumor-Initiating Cells Resistant to SMO Inhibition Elisabeth Peer 1, Sophie Karoline Aichberger 1, Filip Vilotic 1, Wolfgang Gruber 1,†, Thomas Parigger 1,2, Sandra Grund-Gröschke 1,‡, Dominik Patrick Elmer 1 , Florian Rathje 1, Andrea Ramspacher 1,3 , Mirko Zaja 4, Susanne Michel 4, Svetlana Hamm 4 and Fritz Aberger 1,* 1 Department of Bioscience, Cancer Cluster Salzburg, Paris-Lodron University Salzburg, 5020 Salzburg, Austria; [email protected] (E.P.); [email protected] (S.K.A.); viloticfi@web.de (F.V.); gruber.wolfi@gmail.com (W.G.); [email protected] (T.P.); [email protected] (S.G.-G.); [email protected] (D.P.E.); fl[email protected] (F.R.); [email protected] (A.R.) 2 Laboratory for Immunological and Molecular Cancer Research (SCRI-LIMCR), Salzburg Cancer Research Institute, Cancer Cluster Salzburg, IIIrd Medical Department, Paracelsus Medical University Salzburg, 5020 Salzburg, Austria 3 Research Program for Receptor Biochemistry and Tumor Metabolism, Department of Pediatrics, University Hospital of the Paracelsus Medical University, 5020 Salzburg, Austria 4 4SC AG, Planegg-Martinsried, 82152 Planegg, Germany; [email protected] (M.Z.); [email protected] (S.M.); [email protected] (S.H.) Citation: Peer, E.; Aichberger, S.K.; * Correspondence: [email protected]; Tel.: +43-662-8044-5792 Vilotic, F.; Gruber, W.; Parigger, T.; † Present address: EVER Pharma GmbH, 4866 Unterach, Austria. Grund-Gröschke, S.; Elmer, D.P.; ‡ Present address: Immatics Biotechnologies GmbH, 72076 Tübingen, Germany. -

Protein Interactors of the Cellular Tumor Antigen P53 with Response to Ionizing Radiation
IPEN Informe Científico Tecnológico. Volumen 12 (2012). p. 49-50. ISSN 1684-1662 Protein interactors of the cellular tumor antigen p53 with response to ionizing radiation Silvia Vasquez1,3,4,*, Roberto Pineda2, Mohanalatha Chandrasekharan3, Prashanth Suravajhala3,4 1 Dirección de Investigación y Desarrollo, Centro Nuclear RACSO, Instituto Peruano de Energía Nuclear, Av. Canadá 1470, Lima 41, Perú 2 Facultad de Ciencias Biológicas, Universidad Ricardo Palma, Av. Benavides 5440, Surco, Lima 33, Perú 3 Bioclues.org, IKP Knowledge Park, Secunderabad, 500009 AP, India 4 Bioinformatics Organization, Hudson, Massachusetts, United States of America Abstract TP53 interacts with the genes CDKN1A, CSNK1A1, CSNK1D, BAX, ATM, MDM2, HIF1A,SP1. This consensus network has many binding regions involved in phosphorylation. To find whether or not the interactions are involved in ionizing radiation (IR), we used protein interaction mining and visualization tools to decipher the network. We conclude that ATM, PCNA, TP53 and CDKN1A show protein interactions on cellular response to IR. Resumen TP53 interactúa con los genes CDKN1A, CSNK1A1, CSNK1D, BAX, ATM, MDM2, HIF1A y SP1. La red consensual mencionada presenta varias regiones de unión que intervienen durante la fosforilación. Para encontrar si estas interacciones participan o no en la respuesta a la radiación ionizante (IR) analizamos la minería de interacciones proteicas y las herramientas de visualización para descifrar la red. Concluimos que ATM, PCNA, TP53 y CDKN1A presentan interacciones proteicas en la respuesta celular a IR. 1. Introduction proteins could also be better candidates and it would be interesting to run pull down assays The cellular tumor antigen p53[1] participates for these proteins to check for their candidacy in the regulation of the cellular cycle and the in CVD. -

Page 1 Exploring the Understudied Human Kinome For
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.02.022277; this version posted June 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Exploring the understudied human kinome for research and therapeutic opportunities Nienke Moret1,2,*, Changchang Liu1,2,*, Benjamin M. Gyori2, John A. Bachman,2, Albert Steppi2, Rahil Taujale3, Liang-Chin Huang3, Clemens Hug2, Matt Berginski1,4,5, Shawn Gomez1,4,5, Natarajan Kannan,1,3 and Peter K. Sorger1,2,† *These authors contributed equally † Corresponding author 1The NIH Understudied Kinome Consortium 2Laboratory of Systems Pharmacology, Department of Systems Biology, Harvard Program in Therapeutic Science, Harvard Medical School, Boston, Massachusetts 02115, USA 3 Institute of Bioinformatics, University of Georgia, Athens, GA, 30602 USA 4 Department of Pharmacology, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA 5 Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University, Chapel Hill, NC 27599, USA Key Words: kinase, human kinome, kinase inhibitors, drug discovery, cancer, cheminformatics, † Peter Sorger Warren Alpert 432 200 Longwood Avenue Harvard Medical School, Boston MA 02115 [email protected] cc: [email protected] 617-432-6901 ORCID Numbers Peter K. Sorger 0000-0002-3364-1838 Nienke Moret 0000-0001-6038-6863 Changchang Liu 0000-0003-4594-4577 Ben Gyori 0000-0001-9439-5346 John Bachman 0000-0001-6095-2466 Albert Steppi 0000-0001-5871-6245 Page 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.04.02.022277; this version posted June 30, 2020. -

Analysis of DNA Methylation Sites Used for Forensic Age Prediction and Their Correlation with Human Aging
Silva DSBS, et al., J Forensic Leg Investig Sci 2021, 7: 054 DOI: 10.24966/FLIS-733X/100054 HSOA Journal of Forensic, Legal & Investigative Sciences Research Article Analysis of DNA Methylation Introduction DNA samples left behind in crime scenes are often used for Sites used for Forensic Age identification purposes by comparing them to reference samples or to a forensic database. In cases where there is no match, investigators may Prediction and their Correlation turn to alternative approaches using advanced technologies to gain valuable leads on phenotypic traits (externally visible characteristics) with Human Aging of the person who had left the DNA material. Previous studies have found that phenotypes can be predicted by using information obtained from different DNA markers, such as single nucleotide Silva DSBS1* and Karantenislis G2 polymorphisms (SNPs), insertion/deletion polymorphisms (InDel), and epigenetic markers [1-11]. It is well established that epigenetic 1Department of Chemistry, Hofstra University, Hempstead-NY, USA modifications are major regulators in the translation of genotype 2John F Kennedy High School, Bellmore-NY, USA to phenotype. Epigenetic regulation encompasses various levels of gene expression and involves modifications of DNA, histones, RNA and chromatin, with functional consequences for the human Abstract genome [12-14]. DNA methylation is the most well-characterized epigenetic modification. It is usually associated with transcriptional Phenotype-related features, such as age, are linked to repression and it involves the addition of a methyl group (– CH3) in genetic components through regulatory pathways and epigenetic the cytosine of CpG dinucleotides. Differences in DNA methylation modifications, such as DNA methylation, are major regulators in levels in specific regions of the genome can affect the expression of the translation of genotype to phenotype. -

Genetic Insights on Sleep Schedules
Review Genetic insights on sleep schedules: this time, it’s PERsonal 1 1,2 1 S.Y. Christin Chong , Louis J. Pta´ cˇek and Ying-Hui Fu 1 Department of Neurology, University of California San Francisco, San Francisco, CA 94158, USA 2 Howard Hughes Medical Institute, San Francisco, CA 94158, USA The study of circadian rhythms is emerging as a fruitful the genetic basis of circadian behaviors, and this opened opportunity for understanding cellular mechanisms that the floodgate for the study of analogous circadian genes in govern human physiology and behavior, fueled by evi- mammalian model systems [3]. However, are human cir- dence directly linking sleep disorders to genetic muta- cadian behaviors also genetically ‘hard-wired’? tions affecting circadian molecular pathways. Familial Although humans are by nature diurnal, there is a wide advanced sleep-phase disorder (FASPD) is the first rec- spectrum of sleep–wake time preferences in the general ognized Mendelian circadian rhythm trait, and affected population, ranging from ‘morning larks’ to ‘night owls’ [4]. individuals exhibit exceptionally early sleep–wake onset With the advent of high-throughput genotyping technolo- due to altered post-translational regulation of period gies, and the identification of conserved circadian genes, homolog 2 (PER2). Behavioral and cellular circadian mounting evidence now supports the role of genetics in rhythms are analogously affected because the circadian regulating sleep timing [5]. Nevertheless, the identifica- period length of behavior is reduced in the absence of tion of causative genes remains challenging due to complex environmental time cues, and cycle duration of the mo- polygenic interactions, confounding environmental factors, lecular clock is likewise shortened.