Prediction and Characterization of Enzymatic Activities Guided By
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
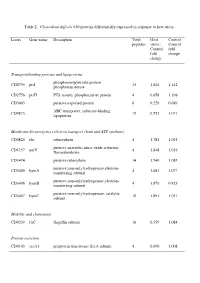
Table 2: Clostridium Difficile 630 Proteins Differentially Expressed in Response to Heat Stress
Table 2: Clostridium difficile 630 proteins differentially expressed in response to heat stress. Locus Gene name Description Total Heat Control : peptides stress : Control Control fold fold change change Transport/binding proteins and lipoproteins phosphoenolpyruvate-protein CD2755 ptsI 15 1.644 1.142 phosphotransferase CD2756 ptsH PTS system, phosphocarrier protein 4 0.658 1.198 CD3043 putative exported protein 6 0.229 0.689 ABC transporter, substrate-binding CD0873 15 0.521 1.071 lipoprotein Membrane bioenergetics (electron transport chain and ATP synthase) CD0825 rbr rubrerythrin 4 1.781 1.035 putative anaerobic nitric oxide reductase CD1157 norV 4 1.848 1.016 flavorubredoxin CD1474 putative ruberythrin 14 1.540 1.085 putative iron-only hydrogenase,electron- CD3405 hymA 4 1.681 1.077 transferring subunit putative iron-only hydrogenase,electron- CD3406 hymB 4 1.870 0.923 transferring subunit putative iron-only hydrogenase, catalytic CD3407 hymC 10 1.891 1.051 subunit Mobility and chemotaxis CD0239 fliC flagellin subunit 16 0.559 1.084 Protein secretion CD0143 secA1 preprotein translocase SecA subunit 4 0.690 1.008 Specific pathways putative oxidoreductase, thiamine diP-binding CD0116 13 1.628 1.121 subunit putative oxidoreductase, acetyl-CoA synthase CD0174 4 2.687 1.055 subunit CD0790 putative NUDIX-family hydrolase 8 1.491 1.32 inosine-uridine preferring nucleoside CD1682 iunH 4 0.518 0.984 hydrolase CD2181 putative aromatic compounds hydrolase 7 0372 0.909 CD2342 sucD succinate-semialdehyde dehydrogenase 6 0.651 0.906 Metabolism -

Generated by SRI International Pathway Tools Version 25.0, Authors S
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_000725805Cyc: Streptomyces xanthophaeus Cellular Overview Connections between pathways are omitted for legibility. -

The Cross-Tissue Metabolic Response of Abalone (Haliotis Midae) to Functional Hypoxia Leonie Venter1, Du Toit Loots1, Lodewyk J
© 2018. Published by The Company of Biologists Ltd | Biology Open (2018) 7, bio031070. doi:10.1242/bio.031070 RESEARCH ARTICLE The cross-tissue metabolic response of abalone (Haliotis midae) to functional hypoxia Leonie Venter1, Du Toit Loots1, Lodewyk J. Mienie1, Peet J. Jansen van Rensburg1, Shayne Mason1, Andre Vosloo2 and Jeremie Z. Lindeque1,* ABSTRACT energetic reserves, resulting in increased oxygen demand beyond the Functional hypoxia is a stress condition caused by the abalone itself as rate of uptake (Morash and Alter, 2016), which limits adenosine a result of increased muscle activity, which generally necessitates the triphosphate (ATP) production via muscle oxidative metabolism employment of anaerobic metabolism if the activity is sustained for (Storey, 2005). During such burst contractile muscle activity, prolonged periods. With that being said, abalone are highly reliant on organisms are generally fuelled by anaerobic metabolism where anaerobic metabolism to provide partial compensation for energy ATP production is made possible from substrate-level production during oxygen-deprived episodes. However, current phosphorylation via the breakdown of phosphagens, but also due to knowledge on the holistic metabolic response for energy metabolism glycolytic degradation of, predominantly, carbohydrates (Baldwin during functional hypoxia, and the contribution of different metabolic and England, 1982; Ellington, 1983). While lipids and proteins are pathways and various abalone tissues towards the overall more ideally used for structural -

Regulation of Haemocyanin Function in Haliotis Iris 255 Also Matched with Adductor Muscle Haemolymph Samples
The Journal of Experimental Biology 205, 253–263 (2002) 253 Printed in Great Britain © The Company of Biologists Limited 2002 JEB3731 The archaeogastropod mollusc Haliotis iris: tissue and blood metabolites and allosteric regulation of haemocyanin function Jane W. Behrens1,3, John P. Elias2,3, H. Harry Taylor3 and Roy E. Weber1,* 1Department of Zoophysiology, Institute Biological Sciences, University of Aarhus, DK 8000 Aarhus, Denmark, 2School of Biological Sciences, Monash University, Clayton, Victoria 3800, Australia and 3Department of Zoology, University of Canterbury, Private Bag 4800, Christchurch, New Zealand *Author for correspondence (e-mail: [email protected]) Accepted 30 October 2001 Summary 2+ 2+ We investigated divalent cation and anaerobic end- Mg and Ca restored the native O2-binding properties product concentrations and the interactive effects of these and the reverse Bohr shift. L- and D-lactate exerted substances and pH on haemocyanin oxygen-binding (Hc- minor modulatory effects on O2-affinity. At in vivo 2+ 2+ O2) in the New Zealand abalone Haliotis iris. During 24 h concentrations of Mg and Ca , the cooperativity is 2+ of environmental hypoxia (emersion), D-lactate and dependent largely on Mg , which modulates the O2 tauropine accumulated in the foot and shell adductor association equilibrium constants of both the high-affinity muscles and in the haemolymph of the aorta, the pedal (KR) and the low-affinity (KT) states (increasing and sinus and adductor muscle lacunae, whereas L-lactate was decreasing, respectively). This allosteric mechanism not detected. Intramuscular and haemolymph D-lactate contrasts with that encountered in other haemocyanins concentrations were similar, but tauropine accumulated and haemoglobins. -

K. ENZYME KINETICS and CATALYSIS Basic Kinetics
K. ENZYME KINETICS AND CATALYSIS Biochemical reactions are too slow to support life at room temperature. Whether it is the synthesis of proteins, the metabolism of sugars or the oxidation of fatty acids, all of it proceeds on a time scale unsuitable for life. Moreover, many reactions that are undesirable proceed on a comparable timescale, making it even more difficult to coordinate the necessary chemistry to support life. How useful would it be to have polynucleotides degrading faster than they are synthesized? The basic reactions associated with the synthesis and degradation of essential polymers, and with the metabolism of nutrients all require massive rate accelerations to be of any service to the cell. By specifically accelerating those reactions of interest without accidentally accelerating undesired reactions, as would happen by simply heating the pot, biochemical catalysts shape chemistry in such a way as to permit every biological process required for survival and propagation. Basic Kinetics Reaction Rates The rate of a reaction is defined as the instantaneous rate of change in the concentration of a starting material (“substrate” in biochemical nomenclature – abbreviated “S”) or a product with respect to time. By definition, reaction rates are always positive, which leads to the following definition of reaction rate, abbreviated v (for velocity) in most of the biochemical literature. S → P (eq. K.1) d[P] d[S] v = = − (eq. K.2) dt dt Because these are instantaneous rates of change, they can be obtained as tangents to curves that plot [P] or [S] vs. time. Typically, one takes the initial rate (Figure K.1) – as close to the point of € initiation of the reaction as possible, since the reaction components are well-defined at that moment. -

Bacterial Selenoproteins: a Role in Pathogenesis and Targets for Antimicrobial Development
University of Central Florida STARS Electronic Theses and Dissertations, 2004-2019 2009 Bacterial Selenoproteins: A Role In Pathogenesis And Targets For Antimicrobial Development Sarah Rosario University of Central Florida Part of the Medical Sciences Commons Find similar works at: https://stars.library.ucf.edu/etd University of Central Florida Libraries http://library.ucf.edu This Doctoral Dissertation (Open Access) is brought to you for free and open access by STARS. It has been accepted for inclusion in Electronic Theses and Dissertations, 2004-2019 by an authorized administrator of STARS. For more information, please contact [email protected]. STARS Citation Rosario, Sarah, "Bacterial Selenoproteins: A Role In Pathogenesis And Targets For Antimicrobial Development" (2009). Electronic Theses and Dissertations, 2004-2019. 3822. https://stars.library.ucf.edu/etd/3822 BACTERIAL SELENOPROTEINS: A ROLE IN PATHOGENESIS AND TARGETS FOR ANTIMICROBIAL DEVELOPMENT. by SARAH E. ROSARIO B.S. Florida State University, 2000 M.P.H. University of South Florida, 2002 A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Burnett School of Biomedical Sciences in the College of Medicine at the University of Central Florida Orlando, Florida Summer Term 2009 Major Professor: William T. Self © 2009 Sarah E. Rosario ii ABSTRACT Selenoproteins are unique proteins in which selenocysteine is inserted into the polypeptide chain by highly specialized translational machinery. They exist within all three kingdoms of life. The functions of these proteins in biology are still being defined. In particular, the importance of selenoproteins in pathogenic microorganisms has received little attention. We first established that a nosocomial pathogen, Clostridium difficile, utilizes a selenoenzyme dependent pathway for energy metabolism. -

Energy Metabolism in the Tropical Abalone, Haliotis Asinina Linné: Comparisons with Temperate Abalone Species ⁎ J
Journal of Experimental Marine Biology and Ecology 342 (2007) 213–225 www.elsevier.com/locate/jembe Energy metabolism in the tropical abalone, Haliotis asinina Linné: Comparisons with temperate abalone species ⁎ J. Baldwin a, , J.P. Elias a, R.M.G. Wells b, D.A. Donovan c a School of Biological Sciences, Monash University, Clayton, Victoria 3800, Australia b School of Biological Sciences, The University of Auckland, Private Bag 92019, Auckland, New Zealand c Department of Biology, MS 9160, Western Washington University, Bellingham, WA 98225, USA Received 15 March 2006; received in revised form 14 July 2006; accepted 12 September 2006 Abstract The abalone, Haliotis asinina, is a large, highly active tropical abalone that feeds at night on shallow coral reefs where oxygen levels of the water may be low and the animals can be exposed to air. It is capable of more prolonged and rapid exercise than has been reported for temperate abalone. These unusual behaviours raised the question of whether H. asinina possesses enhanced capacities for aerobic or anaerobic metabolism. The blood oxygen transport system of H. asinina resembles that of temperate abalone in terms of a large hemolymph volume, similar hemocyanin concentrations, and in most hemocyanin oxygen binding properties; however, absence of a Root effect appears confined to hemocyanin from H. asinina and may assist oxygen uptake when hemolymph pH falls during exercise or environmental hypoxia. During exposure to air, H. asinina reduces oxygen uptake by at least 20-fold relative to animals at rest in aerated seawater, and there is no significant ATP production from anaerobic glycolysis or phosphagen hydrolysis in the foot or adductor muscles. -

Designed Mono- and Di-Covalent Inhibitors Trap Modeled Functional Motions for Trypanosoma Cruzi Proline Racemase in Crystallography
RESEARCH ARTICLE Designed mono- and di-covalent inhibitors trap modeled functional motions for Trypanosoma cruzi proline racemase in crystallography 1¤a 2¤b 2 Patricia de Aguiar Amaral , Delphine Autheman , Guilherme Dias de MeloID , 1 1 2¤c 2¤d Nicolas Gouault , Jean-FrancËois Cupif , Sophie GoyardID , Patricia Dutra , 2 2 3 4 4 Nicolas Coatnoan , Alain Cosson , Damien Monet , Frederick Saul , Ahmed HaouzID , 1 3 2¤e a1111111111 Philippe Uriac *, Arnaud Blondel *, Paola MinoprioID * a1111111111 a1111111111 1 Universite de Rennes 1, Equipe Chimie organique et interfaces (CORINT), UMR 6226 Sciences Chimiques de Rennes, Rennes, France, 2 Institut Pasteur, Laboratoire des Processus Infectieux à TrypanosomatideÂs, a1111111111 DeÂpartement Infection et EpideÂmiologie, Paris, France, 3 Institut Pasteur, Unite de Bioinformatique a1111111111 Structurale, DeÂpartement de Biologie Structurale et Chimie, CNRS-UMR 3528, Paris, France, 4 Institut Pasteur, Plateforme de Cristallographie, DeÂpartement de Biologie Structurale et Chimie, CNRS-UMR 3528, Paris, France ¤a Current address: LaPlaM/PPGCA, Universidade do Extremo Sul Catarinense, CriciuÂmaÐSC, Brazil ¤b Current address: Cell Surface Signalling Laboratory, Wellcome Trust Sanger Institute, Wellcome Trust OPEN ACCESS Genome Campus, Cambridge, United Kingdom Citation: Amaral PdA, Autheman D, de Melo GD, ¤c Current address: Institut Pasteur, Centre d'Innovation et Recherche Technologique, Paris, France Gouault N, Cupif J-F, Goyard S, et al. (2018) ¤d Current address: LaboratoÂrio de BioquõÂmica de ProtozoaÂrios e Imunofisiologia do ExercõÂcio, Departamento de Microbiologia, Imunologia e Parasitologia, Universidade do Estado do Rio de Janeiro, Rio Designed mono- and di-covalent inhibitors trap de Janeiro, Brazil modeled functional motions for Trypanosoma cruzi ¤e Current address: Trypanosomatids Infectious Processes Laboratory, Scientific Platform Pasteur-USP, proline racemase in crystallography. -

Discovery of a Novel Amino Acid Racemase Through Exploration of Natural Variation in Arabidopsis Thaliana
Discovery of a novel amino acid racemase through exploration of natural variation in Arabidopsis thaliana Renee C. Straucha,b, Elisabeth Svedinc, Brian Dilkesc, Clint Chappled, and Xu Lia,b,1 aPlants for Human Health Institute, North Carolina State University, Kannapolis, NC 28081; bDepartment of Plant and Microbial Biology, North Carolina State University, Raleigh, NC 27695; cDepartment of Horticulture and Landscape Architecture, Purdue University, West Lafayette, IN 47907; and dDepartment of Biochemistry, Purdue University, West Lafayette, IN 47907 Edited by Justin O. Borevitz, Australian National University, Canberra, ACT, Australia, and accepted by the Editorial Board August 1, 2015 (received for review February 16, 2015) Plants produce diverse low-molecular-weight compounds via spe- contribution to the variation in at least three-fourths of detected cialized metabolism. Discovery of the pathways underlying produc- mass peaks (8). tion of these metabolites is an important challenge for harnessing Here we describe an integrated transdisciplinary platform, the huge chemical diversity and catalytic potential in the plant king- combining metabolomics, genetics, and genomics, to exploit the dom for human uses, but this effort is often encumbered by the biochemical and genetic diversity of natural accessions of the necessity to initially identify compounds of interest or purify a cata- model plant A. thaliana to uncover associations between genes lyst involved in their synthesis. As an alternative approach, we have and metabolites. Using this platform, we linked a differentially performed untargeted metabolite profiling and genome-wide asso- accumulating metabolite, identified through chemical analysis as ciation analysis on 440 natural accessions of Arabidopsis thaliana. N-malonyl-D-allo-isoleucine (NMD-Ile), to a previously unchar- This approach allowed us to establish genetic linkages between acterized gene identified as an amino acid racemase through metabolites and genes. -

(12) United States Patent (10) Patent No.: US 9,695.451 B2 Chen Et Al
US0096.95451B2 (12) United States Patent (10) Patent No.: US 9,695.451 B2 Chen et al. (45) Date of Patent: *Jul. 4, 2017 (54) POLYNUCLEOTIDES ENCODING (52) U.S. Cl. ENGINEERED IMINE REDUCTASES CPC ............ CI2P 13/02 (2013.01); C12N 9/0028 (2013.01); CI2P 13/001 (2013.01): CI2P (71) Applicant: CODEXIS, INC., Redwood City, CA 13/04 (2013.01); C12P 13/06 (2013.01); CI2P (US) 17/10 (2013.01); C12P 17/12 (2013.01); CI2P (72) Inventors: 'E'O O he, By- - - SN R J. 17/165CI2P (2013.01); 17/188 (2013.01);CI2P 17/185 C12Y (2013.01); 105/01 NESCO.ARS y (2013.01); C12Y 105/01023 (2013.01); C12Y Sukumaran, Singapore (SG), Derek 105/01024 (2013.01); C12Y 105/01028 Smith, Singapore (SG); Jeffrey C. (2013.01); Y02P 20/52 (2015.11) Moore, Westfield, NJ (US); Gregory (58) Field of Classification Search Hughes, Scotch Plains, NJ (US); Jacob CPC ..... Y02P 20/52; C12N 9/0028; C12N 9/0016: Janey, New York, NY (US); Gjalt W. C12P 13/001; C12P 17/10; C12P 13/02: Huisman, Redwood City, CA (US); C12P 13/04; C12P 13/06; C12P 17/12: Scott J. Novick, Palo Alto, CA (US); C12P 17/165; C12P 17/185; C12P Nicholas J. Agard, San Francisco, CA 17/188; C12P 21/02: C12Y 105/01028; (US); Oscar Alvizo, Fremont, CA (US); C12Y 105/01, C12Y 105/01023; C12Y it's AS Ms. Park, Sé 105/01024; C07K 14/61 Stefanie; Wan Ng Lin Minor, Yeo, RedwoodS1ngapore City, CA See application file for complete search history. -

NIH Public Access Author Manuscript Curr Opin Chem Biol
NIH Public Access Author Manuscript Curr Opin Chem Biol. Author manuscript; available in PMC 2010 October 1. NIH-PA Author ManuscriptPublished NIH-PA Author Manuscript in final edited NIH-PA Author Manuscript form as: Curr Opin Chem Biol. 2009 October ; 13(4): 468±476. doi:10.1016/j.cbpa.2009.06.023. Pyridoxal 5'-Phosphate: Electrophilic Catalyst Extraordinaire John P. Richard†,*, Tina L. Amyes†, Juan Crugeiras#, and Ana Rios# †Department of Chemistry, University at Buffalo, SUNY, Buffalo, NY 14260-3000, USA. #Departamento de Química Física, Facultad de Química, Universidad de Santiago, 15782 Santiago de Compostela, Spain. Abstract Studies of nonenzymatic electrophilic catalysis of carbon deprotonation of glycine show that pyridoxal 5'-phosphate (PLP) strongly enhances the carbon acidity of α-amino acids, but that this is not the overriding mechanistic imperative for cofactor catalysis. Although the fully protonated PLP-glycine iminium ion adduct exhibits an extraordinary low α-imino carbon acidity (pKa = 6), the more weakly acidic zwitterionic iminium ion adduct (pKa = 17) is selected for use in enzymatic reactions. The similar α-imino carbon acidities of the iminium ion adducts of glycine with 5'- deoxypyridoxal and with phenylglyoxylate shows that the cofactor pyridine nitrogen plays a relatively minor role in carbanion stabilization. The 5'-phosphodianion group of PLP likely plays an important role in catalysis by providing up to 12 kcal/mol of binding energy that may be utilized for transition state stabilization. Introduction Scientists prize the rush of adrenalin that comes with making an original discovery, or with bringing order to seemingly disconnected experimental observations. Snell and Braunstein must have received tremendous satisfaction from their independent discovery, more than sixty years ago, that heating pyridoxal 5'-phosphate (PLP) with an amino acid yields the products of transamination of the amino acid [1]. -

SI Appendix Index 1
SI Appendix Index Calculating chemical attributes using EC-BLAST ................................................................................ 2 Chemical attributes in isomerase reactions ............................................................................................ 3 Bond changes …..................................................................................................................................... 3 Reaction centres …................................................................................................................................. 5 Substrates and products …..................................................................................................................... 6 Comparative analysis …........................................................................................................................ 7 Racemases and epimerases (EC 5.1) ….................................................................................................. 7 Intramolecular oxidoreductases (EC 5.3) …........................................................................................... 8 Intramolecular transferases (EC 5.4) ….................................................................................................. 9 Supporting references …....................................................................................................................... 10 Fig. S1. Overview …............................................................................................................................