(CD-P-PH/PHO) Report Classification/Justifica
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
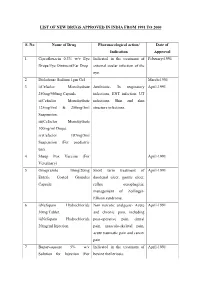
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

Appendix a Common Abbreviations Used in Medication
UNIVERSITY OF AMSTERDAM MASTERS THESIS Impact of Medication Grouping on Fall Risk Prediction in Elders: A Retrospective Analysis of MIMIC-III Critical Care Database Student: SRP Mentor: Noman Dormosh Dr. Martijn C. Schut Student No. 11412682 – SRP Tutor: Prof. dr. Ameen Abu-Hanna SRP Address: Amsterdam University Medical Center - Location AMC Department Medical Informatics Meibergdreef 9, 1105 AZ Amsterdam Practice teaching period: November 2018 - June 2019 A thesis submitted in fulfillment of the requirements for the degree of Master of Medical Informatics iii Abstract Background: Falls are the leading cause of injury in elderly patients. Risk factors for falls in- cluding among others history of falls, old age, and female gender. Research studies have also linked certain medications with an increased risk of fall in what is called fall-risk-increasing drugs (FRIDs), such as psychotropics and cardiovascular drugs. However, there is a lack of consistency in the definitions of FRIDs between the studies and many studies did not use any systematic classification for medications. Objective: The aim of this study was to investigate the effect of grouping medications at different levels of granularity of a medication classification system on the performance of fall risk prediction models. Methods: This is a retrospective analysis of the MIMIC-III cohort database. We created seven prediction models including demographic, comorbidity and medication variables. Medica- tions were grouped using the anatomical therapeutic chemical classification system (ATC) starting from the most specific scope of medications and moving up to the more generic groups: one model used individual medications (ATC level 5), four models used medication grouping at levels one, two, three and four of the ATC and one model did not include med- ications. -

Nizatidine) Oral Solution
Axid® (nizatidine) Oral Solution Description: Nizatidine (USP) is a histamine H2-receptor antagonist. Chemically, it is N-[2-[[[2-[(dimethylamino)methyl]-4-thiazolyl]methyl]thio]ethyl]-N'-methyl-2-nitro-1,1- ethenediamine. The structural formula is as follows: Nizatidine has the empirical formula C12H21N5O2S2 representing a molecular weight of 331.47. It is an off-white to buff crystalline solid that is soluble in water. Nizatidine has a bitter taste and mild sulfur-like odor. Axid® Oral Solution is formulated as a clear, yellow, oral solution with bubble gum flavor and each 1 mL contains 15 mg of nizatidine. Axid® Oral Solution also contains the inactive ingredients methylparaben, propylparaben, glycerin, sodium alginate, purified water, sodium chloride, saccharin sodium, sodium citrate dihydrate, citric acid anhydrous, sucrose, bubble gum flavor, artificial sweetness enhancer, and sodium hydroxide. Clinical Pharmacology in Adults: Nizatidine is a competitive, reversible inhibitor of histamine at the histamine H2-receptors, particularly those in the gastric parietal cells. Antisecretory Activity—1. Effects on Acid Secretion: Nizatidine significantly inhibited nocturnal gastric acid secretion for up to 12 hours. Nizatidine also significantly inhibited gastric acid secretion stimulated by food, caffeine, betazole, and pentagastrin (Table 1). Table 1. Effect of Oral Nizatidine on Gastric Acid Secretion % Inhibition of Gastric Acid Output by Dose (mg) Time After Dose (h) 20-50 75 100 150 300 Nocturnal Up to 10 57 - 73 - 90 Betazole Up to 3 - 93 - 100 99 Pentagon Up to 6 - 25 - 64 67 Meal Up to 4 41 64 - 98 97 Caffeine Up to 3 - 73 - 85 96 2. Effects on Other Gastrointestinal Secretions—Pepsin: Oral administration of 75 to 300 mg of nizatidine did not affect pepsin activity in gastric secretions. -

Product List March 2019 - Page 1 of 53
Wessex has been sourcing and supplying active substances to medicine manufacturers since its incorporation in 1994. We supply from known, trusted partners working to full cGMP and with full regulatory support. Please contact us for details of the following products. Product CAS No. ( R)-2-Methyl-CBS-oxazaborolidine 112022-83-0 (-) (1R) Menthyl Chloroformate 14602-86-9 (+)-Sotalol Hydrochloride 959-24-0 (2R)-2-[(4-Ethyl-2, 3-dioxopiperazinyl) carbonylamino]-2-phenylacetic 63422-71-9 acid (2R)-2-[(4-Ethyl-2-3-dioxopiperazinyl) carbonylamino]-2-(4- 62893-24-7 hydroxyphenyl) acetic acid (r)-(+)-α-Lipoic Acid 1200-22-2 (S)-1-(2-Chloroacetyl) pyrrolidine-2-carbonitrile 207557-35-5 1,1'-Carbonyl diimidazole 530-62-1 1,3-Cyclohexanedione 504-02-9 1-[2-amino-1-(4-methoxyphenyl) ethyl] cyclohexanol acetate 839705-03-2 1-[2-Amino-1-(4-methoxyphenyl) ethyl] cyclohexanol Hydrochloride 130198-05-9 1-[Cyano-(4-methoxyphenyl) methyl] cyclohexanol 93413-76-4 1-Chloroethyl-4-nitrophenyl carbonate 101623-69-2 2-(2-Aminothiazol-4-yl) acetic acid Hydrochloride 66659-20-9 2-(4-Nitrophenyl)ethanamine Hydrochloride 29968-78-3 2,4 Dichlorobenzyl Alcohol (2,4 DCBA) 1777-82-8 2,6-Dichlorophenol 87-65-0 2.6 Diamino Pyridine 136-40-3 2-Aminoheptane Sulfate 6411-75-2 2-Ethylhexanoyl Chloride 760-67-8 2-Ethylhexyl Chloroformate 24468-13-1 2-Isopropyl-4-(N-methylaminomethyl) thiazole Hydrochloride 908591-25-3 4,4,4-Trifluoro-1-(4-methylphenyl)-1,3-butane dione 720-94-5 4,5,6,7-Tetrahydrothieno[3,2,c] pyridine Hydrochloride 28783-41-7 4-Chloro-N-methyl-piperidine 5570-77-4 -

Impact of Itopride and Domperidone on Sensitivity of Gastric Distention and Gastric Accommodation in Healthy Volunteers
Impact of Itopride and Domperidone on sensitivity of gastric distention and gastric accommodation in healthy volunteers. Karen Van den Houte, Florencia Carbone, Ans Pauwels, Rita Vos, Tim Vanuytsel, Jan Tack Translational Research Center for Gastrointestinal Disorders, KULeuven, Belgium BACKGROUND & AIM RESULTS Functional dyspepsia (FD), defined as upper abdominal symptoms affecting Conduct of the study Gastric accommodation daily life, as postprandial fullness, early satiation, epigastric pain, and epigastric • 15 healthy volunteers (9 female, 6 male) • No significant differences in VAS scores before and after meal burning, without any underlying organic disease, is one of the most common • Mean age: 28.3±5.8 years ingestion and no significant differences in preprandial intragastric functional gastrointestinal disorders (1). volumes were observed. Itopride, a prokinetic drug with dopamine D2-antagonistic and cholinesterase Gastric compliance and gastric sensitivity • Postprandial gastric volumes and gastric accommodation were inhibitor properties, is frequently used to treat functional dyspepsia. Its effects I50, I100, and D10 did not affect fasting or postprandial gastric significantly lower for I50 and for D10, compared to placebo. on gastric sensitivity and accommodation are unknown (2), compliance and gastric sensitivity to distention significantly The aim of this study is to evaluate the effect of Itopride, compared to compared to placebo. Domperidone, a dopamine D2 receptor antagonist, on the sensitivity to gastric * Placebo balloon -

Carboxylic Acid Derivates
Europaisches Patentamt 0 367 484 J> European Patent Office CO Publication number: A1 Office europeen des brevets © EUROPEAN PATENT APPLICATION A61K 31/29 © Application number: 89310994.2 © int. ci.5: C07D 233/64 , C07D 277/42 C07D 277/28 @ Date of filing: 25.10.89 C07D 295/08 C07D 249/14 C07D 211/20 C07D 417/04 © Priority: 26.10.88 GB 8825058 © Applicant: GLAXO GROUP LIMITED 26.06.89 GB 8914631 Clarges House 6-12 Clarges Street London W1Y8DH(GB) © Date of publication of application: 09.05.90 Bulletin 90/19 © Inventor: Clitherow, John Watson 54, Gilders © Designated Contracting States: Sawbridgeworth Hertfordshire(GB) AT BE CH DE ES FR GB GR IT LI LU NL SE © Representative: Marchant, James Ian et al Elkington and Fife Beacon House 113 Kingsway London WC2B 6PP(GB) © Carboxylic acid derivates. © The invention relates to salts formed between basic H2-receptor antagonists and a complex of bismuth with a carboxylic acid, and solvate of such salts, excluding salts in which the basic H2-receptor antagonist is ranitidine. Examples of suitable carboxylic acids are citric acid and tartaric acid. Examples of basic rVreceptor antagonists are cimetidine, sufotidine famotidine and nizatidine. The salts ares useful in the treatment of gastrointestinal disorders, particularly gastroduodenal conditions. The salts show the antisecretory activity associated with the basic H2-receptor antagonist together with antibacterial activity against Campylobacter pylori and they also possess cytoprotective properties. 00 (0 a. LU Xerox Copy Centre EP 0 367 484 A1 CARBOXYLIC ACID DERIVATIVES This invention relates to salts of compounds having antagonist activity at histamine H2- receptors, to a process for the preparation thereof, to pharmaceutical compositions containing them and to their use in therapeutics. -

The In¯Uence of Medication on Erectile Function
International Journal of Impotence Research (1997) 9, 17±26 ß 1997 Stockton Press All rights reserved 0955-9930/97 $12.00 The in¯uence of medication on erectile function W Meinhardt1, RF Kropman2, P Vermeij3, AAB Lycklama aÁ Nijeholt4 and J Zwartendijk4 1Department of Urology, Netherlands Cancer Institute/Antoni van Leeuwenhoek Hospital, Plesmanlaan 121, 1066 CX Amsterdam, The Netherlands; 2Department of Urology, Leyenburg Hospital, Leyweg 275, 2545 CH The Hague, The Netherlands; 3Pharmacy; and 4Department of Urology, Leiden University Hospital, P.O. Box 9600, 2300 RC Leiden, The Netherlands Keywords: impotence; side-effect; antipsychotic; antihypertensive; physiology; erectile function Introduction stopped their antihypertensive treatment over a ®ve year period, because of side-effects on sexual function.5 In the drug registration procedures sexual Several physiological mechanisms are involved in function is not a major issue. This means that erectile function. A negative in¯uence of prescrip- knowledge of the problem is mainly dependent on tion-drugs on these mechanisms will not always case reports and the lists from side effect registries.6±8 come to the attention of the clinician, whereas a Another way of looking at the problem is drug causing priapism will rarely escape the atten- combining available data on mechanisms of action tion. of drugs with the knowledge of the physiological When erectile function is in¯uenced in a negative mechanisms involved in erectile function. The way compensation may occur. For example, age- advantage of this approach is that remedies may related penile sensory disorders may be compen- evolve from it. sated for by extra stimulation.1 Diminished in¯ux of In this paper we will discuss the subject in the blood will lead to a slower onset of the erection, but following order: may be accepted. -

Comparative Effects of Cimetidine and Famotidine on the Vagally Stimulated Acid Secretion in the Isolated Mouse Whole Stomach
Comparative Effects of Cimetidine and Famotidine on the Vagally Stimulated Acid Secretion in the Isolated Mouse Whole Stomach Kazuo Watanabe1, Shingo Yano1, Masayuki Yamamoto1 and Shoko Kanaoka2 1Laboratory of Chemical Pharmacology, Department of Drug Evaluation and Toxicological Sciences, Faculty of Pharmaceutical Sciences, Chiba University, 1-33, Yayoi-cho, Inage-ku, Chiba 263, Japan 2Research Institute for Wakan-Yaku, Toyama Medical and Pharmaceutical University, Toyama 930-01, Japan Received July 15, 1992 Accepted December 10, 1992 ABSTRACT-We investigated the effects of cimetidine and famotidine on the acid secretory response to elec trical vagal stimulation, bethanechol and histamine in the isolated mouse whole stomach preparation. The acid secretion elicited by electrical vagal stimulation at the position of the esophagus (10 Hz, 0.3 msec, 10 V for 5 min) was reproducible by repeated stimulation in each preparation, and it was abolished by tetrodo toxin, atropine and hexamethonium. This vagally stimulated acid secretion was abolished by cimetidine (3 mM), while it was only partly inhibited by famotidine (10-100 ƒÊM). Histamine (100 ƒÊM)-induced acid secre tion was inhibited by cimetidine and famotidine, and the doses of these drugs required for complete inhibi tion were 3 mM and 10 ƒÊM, respectively. In contrast, bethanechol (10 ƒÊM)-induced acid secretion was slight ly reduced by famotidine (1-100 ƒÊM), but markedly reduced by cimetidine (3 mM). In the guinea pig ileum, millimolar concentrations of cimetidine and famotidine shifted the dose-response curve of the contractile response to acetylcholine rightward. These findings suggest that the inhibitory effect of cimetidine on the vagally stimulated or bethanechol-induced acid secretion is elicited at least partly through mechanisms different from H2-antagonism. -

A Comparative Evaluation of Lafutidine and 2 Rabeprazole in The
1 *Original research paper 2 A comparative evaluation of Lafutidine and 3 Rabeprazole in the treatment of gastritis and 4 peptic ulcer: A double-blind, randomized study 5 in Indian patients. 6 Dr. Sanjay Kumar 1, Dr. Bhupesh Dewan 2*, Deepashri Shah 2 7 8 1Global Liver and Gastroenterology Centre, Bhopal, India 2 9 Medical Department, Zuventus Healthcare Ltd., Mumbai, India 10 11 . 12 ABSTRACT 13 Aims: To assess the efficacy of lafutidine therapy versus rabeprazole in Indian patients with endoscopically and histologically proven gastritis and peptic ulcer. Study design: A double blind, double dummy, randomized, comparative study. Place and Duration of Study: Global Liver and Gastroenterology Centre, Bhopal, India, between March 2010 and October 2010. Methodology: A total of 100 patients were enrolled, including 50 with endoscopically and histologically proven gastritis and other 50 with peptic ulcer (over 5 mm in diameter). Each group was randomized to receive either lafutidine or rabeprazole tablet and their corresponding competitor placebo dummy tablet, for a period of 4 weeks. Gastritis/ulcer cure rates confirmed by endoscopic histology, symptom response and Helicobacter pylori (H. Pylori) eradication were compared among the two drugs Results: Complete cure of gastritis was observed in all the patients (100%) treated with lafutidine and 95.24% [20/21; 95% CI: 76.18 to 99.88%] patients treated with rabeprazole. Complete cure of ulcer was observed in 72.0% (18/25, 95% CI = 50.61 to 87.93%) and 79.16% (19/24, 95% CI = 57.85 to 92.87%) patients treated with lafutidine and rabeprazole respectively. There was no significant difference in gastritis/ulcer cure rate and symptom response rate between the two treatment groups at the end of the study. -

Nexium Control, Esomeprazole
27 June 2013 EMA/498929/2013 Committee for Medicinal Products for Human Use (CHMP) Assessment report Nexium Control International non-proprietary name: esomeprazole Procedure No. EMEA/H/C/002618 Note Assessment report as adopted by the CHMP with all information of a commercially confidential nature deleted. 7 Westferry Circus ● Canary Wharf ● London E14 4HB ● United Kingdom Telephone +44 (0)20 7418 8400 Facsimile +44 (0)20 7418 8613 E -mail [email protected] Website www.ema.europa.eu An agency of the European Union © European Medicines Agency, 2013. Reproduction is authorised provided the source is acknowledged. Product information Marketing authorisation application Name of the medicinal product: Nexium Control Applicant: AstraZeneca AB Building 411A, Floor 4 S - 151 85 Södertälje SWEDEN Active substance: esomeprazole (as magnesium trihydrate) International Nonproprietary Name/Common Name: esomeprazole Pharmaco-therapeutic group Proton pump inhibitors (ATC Code): (A02BC05) Therapeutic indication: Nexium Control is indicated for the short-term treatment of reflux symptoms (e.g. heartburn and acid regurgitation) in adults. Pharmaceutical form: Gastro-resistant tablet Strength: 20 mg Route of administration: Oral use Packaging: blister (PVC/PVDC) Package sizes: 7 tablets and 14 tablets Assessment report EMA/498929/2013 Page 2/70 Table of contents 1. Background information on the procedure .............................................. 6 1.1. Submission of the dossier ..................................................................................... -

Ijcep0048253.Pdf
Int J Clin Exp Pathol 2017;10(4):4089-4098 www.ijcep.com /ISSN:1936-2625/IJCEP0048253 Original Article Comparative effectiveness of different recommended doses of omeprazole and lansoprazole for gastroesophageal reflux disease: a meta-analysis of published data Feng Liu1, Jing Wang1, Hailong Wu1, Hui Wang2, Jianxiang Wang1, Rui Zhou1, Zhi Zhu3 Departments of 1Surgery, 2Gastroenterology, 3Epidemiology and Biostatistics, Puai Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China Received January 6, 2017; Accepted February 20, 2017; Epub April 1, 2017; Published April 15, 2017 Abstract: This meta-analysis aims to evaluate the effectiveness of different recommended doses of omeprazole and lansoprazole on gastroesophageal reflux diseases (GERD) in adults. The electronic databases of PubMed, EMBASE, Cochrane Library, and ClinicalTrials.gov were searched before September 13 2016. Fifteen eligible studies were identified involving 8752 patients in our meta-analysis. For the healing outcome of esophagitis, compared with 15 mg per day of lansoprazole, there were significant difference of both 30 mg per day of lansoprazole (RR=1.29, 95% CI [1.01, 1.66], I2=79.3%, P=0.028) and 60 mg per day of lansoprazole (RR=1.59, 95% CI [1.28, 1.99], I2=Not ap- plicable (NA), P=NA), and the other result were not significantly different between 60 mg per day and 30 mg per day. For relief of symptoms, our result indicated a significant difference between 20 mg per day and 10 mg per day of omeprazole (RR=1.21, 95% CI [1.06, 1.39], I2=53.9%, P=0.089); the overall result indicated a significant difference between lansoprazole and omeprazole (RR=0.93, 95% CI [0.86, 0.999], I2=60.7%, P=0.038). -

Protonix Protonix
NDA 22-020 Page 4 PROTONIX® (pantoprazole sodium) Delayed-Release Tablets PROTONIX® (pantoprazole sodium) For Delayed-Release Oral Suspension Rx only DESCRIPTION ® The active ingredient in PROTONIX (pantoprazole sodium) Delayed-Release Tablets and ® PROTONIX (pantoprazole sodium) For Delayed-Release Oral Suspension is a substituted benzimidazole, sodium 5-(difluoromethoxy)-2-[[(3,4-dimethoxy-2-pyridinyl)methyl] sulfinyl]- 1H-benzimidazole sesquihydrate, a compound that inhibits gastric acid secretion. Its empirical formula is C16H14F2N3NaO4S x 1.5 H2O, with a molecular weight of 432.4. The structural formula is: Pantoprazole sodium sesquihydrate is a white to off-white crystalline powder and is racemic. Pantoprazole has weakly basic and acidic properties. Pantoprazole sodium sesquihydrate is freely soluble in water, very slightly soluble in phosphate buffer at pH 7.4, and practically insoluble in n-hexane. The stability of the compound in aqueous solution is pH-dependent. The rate of degradation increases with decreasing pH. At ambient temperature, the degradation half-life is approximately 2.8 hours at pH 5.0 and approximately 220 hours at pH 7.8. PROTONIX (pantoprazole sodium) is supplied as a delayed-release tablet for oral administration, available in 2 strengths (40 mg and 20 mg); and as delayed-release granules for oral administration, available in the 40 mg strength. NDA 22-020 Page 5 Each PROTONIX (pantoprazole sodium) Delayed-Release tablet contains 45.1 mg or 22.6 mg of pantoprazole sodium sesquihydrate (equivalent to 40 mg or 20 mg pantoprazole, respectively) with the following inactive ingredients: calcium stearate, crospovidone, hypromellose, iron oxide, mannitol, methacrylic acid copolymer, polysorbate 80, povidone, propylene glycol, sodium carbonate, sodium lauryl sulfate, titanium dioxide, and triethyl citrate.