Bone Marrow Leukemia Non- Patient Name Report Date Lymphocytic Acute Myelocytic (AML)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Transcriptional Control of Tissue-Resident Memory T Cell Generation
Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate School of Arts and Sciences COLUMBIA UNIVERSITY 2019 © 2019 Filip Cvetkovski All rights reserved ABSTRACT Transcriptional control of tissue-resident memory T cell generation Filip Cvetkovski Tissue-resident memory T cells (TRM) are a non-circulating subset of memory that are maintained at sites of pathogen entry and mediate optimal protection against reinfection. Lung TRM can be generated in response to respiratory infection or vaccination, however, the molecular pathways involved in CD4+TRM establishment have not been defined. Here, we performed transcriptional profiling of influenza-specific lung CD4+TRM following influenza infection to identify pathways implicated in CD4+TRM generation and homeostasis. Lung CD4+TRM displayed a unique transcriptional profile distinct from spleen memory, including up-regulation of a gene network induced by the transcription factor IRF4, a known regulator of effector T cell differentiation. In addition, the gene expression profile of lung CD4+TRM was enriched in gene sets previously described in tissue-resident regulatory T cells. Up-regulation of immunomodulatory molecules such as CTLA-4, PD-1, and ICOS, suggested a potential regulatory role for CD4+TRM in tissues. Using loss-of-function genetic experiments in mice, we demonstrate that IRF4 is required for the generation of lung-localized pathogen-specific effector CD4+T cells during acute influenza infection. Influenza-specific IRF4−/− T cells failed to fully express CD44, and maintained high levels of CD62L compared to wild type, suggesting a defect in complete differentiation into lung-tropic effector T cells. -

Maintenance of Mammary Epithelial Phenotype by Transcription Factor Runx1 Through Mitotic Gene Bookmarking Joshua Rose University of Vermont
University of Vermont ScholarWorks @ UVM Graduate College Dissertations and Theses Dissertations and Theses 2019 Maintenance Of Mammary Epithelial Phenotype By Transcription Factor Runx1 Through Mitotic Gene Bookmarking Joshua Rose University of Vermont Follow this and additional works at: https://scholarworks.uvm.edu/graddis Part of the Biochemistry Commons, and the Genetics and Genomics Commons Recommended Citation Rose, Joshua, "Maintenance Of Mammary Epithelial Phenotype By Transcription Factor Runx1 Through Mitotic Gene Bookmarking" (2019). Graduate College Dissertations and Theses. 998. https://scholarworks.uvm.edu/graddis/998 This Thesis is brought to you for free and open access by the Dissertations and Theses at ScholarWorks @ UVM. It has been accepted for inclusion in Graduate College Dissertations and Theses by an authorized administrator of ScholarWorks @ UVM. For more information, please contact [email protected]. MAINTENANCE OF MAMMARY EPITHELIAL PHENOTYPE BY TRANSCRIPTION FACTOR RUNX1 THROUGH MITOTIC GENE BOOKMARKING A Thesis Presented by Joshua Rose to The Faculty of the Graduate College of The University of Vermont In Partial Fulfillment of the Requirements for the Degree of Master of Science Specializing in Cellular, Molecular, and Biomedical Sciences January, 2019 Defense Date: November 12, 2018 Thesis Examination Committee: Sayyed Kaleem Zaidi, Ph.D., Advisor Gary Stein, Ph.D., Advisor Seth Frietze, Ph.D., Chairperson Janet Stein, Ph.D. Jonathan Gordon, Ph.D. Cynthia J. Forehand, Ph.D. Dean of the Graduate College ABSTRACT Breast cancer arises from a series of acquired mutations that disrupt normal mammary epithelial homeostasis and create multi-potent cancer stem cells that can differentiate into clinically distinct breast cancer subtypes. Despite improved therapies and advances in early detection, breast cancer remains the leading diagnosed cancer in women. -

3. Inflammasomes
UNIVERSIDAD DE MURCIA ESCUELA INTERNACIONAL DE DOCTORADO Characterization of Caiap and Wdr90 as Novel Inflammasome Components Involved in the Resistance to Salmonella enterica serovar Typhimurium. Caracterización de Caiap y Wdr90 como Nuevos Componentes del Inflamasoma Implicados en la Resistencia a Salmonella enterica serovar Typhimurium Dña. Ana Valera Pérez 2018 TABLE OF CONTENTS ABBREVIATIONS ................................................................................................................ 9 SUMMARY....................................................................................................................... 19 INTRODUCTION .............................................................................................................. 23 1. The immune system .............................................................................. 25 1.1. The innate immune system in fish ....................................................... 26 1.2. Adaptive immune system in fish .......................................................... 29 2. Immune system and inflammasomes ................................................... 30 2.1. PRRs ................................................................................................... 31 2.2. Toll like receptors .............................................................................. 32 2.3. NOD-like receptors ............................................................................ 32 3. Inflammasomes .................................................................................... -

The Genetic Program of Pancreatic Beta-Cell Replication in Vivo
Page 1 of 65 Diabetes The genetic program of pancreatic beta-cell replication in vivo Agnes Klochendler1, Inbal Caspi2, Noa Corem1, Maya Moran3, Oriel Friedlich1, Sharona Elgavish4, Yuval Nevo4, Aharon Helman1, Benjamin Glaser5, Amir Eden3, Shalev Itzkovitz2, Yuval Dor1,* 1Department of Developmental Biology and Cancer Research, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel. 3Department of Cell and Developmental Biology, The Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel 4Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah, The Institute for Medical Research Israel- Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 5Endocrinology and Metabolism Service, Department of Internal Medicine, Hadassah-Hebrew University Medical Center, Jerusalem 91120, Israel *Correspondence: [email protected] Running title: The genetic program of pancreatic β-cell replication 1 Diabetes Publish Ahead of Print, published online March 18, 2016 Diabetes Page 2 of 65 Abstract The molecular program underlying infrequent replication of pancreatic beta- cells remains largely inaccessible. Using transgenic mice expressing GFP in cycling cells we sorted live, replicating beta-cells and determined their transcriptome. Replicating beta-cells upregulate hundreds of proliferation- related genes, along with many novel putative cell cycle components. Strikingly, genes involved in beta-cell functions, namely glucose sensing and insulin secretion were repressed. Further studies using single molecule RNA in situ hybridization revealed that in fact, replicating beta-cells double the amount of RNA for most genes, but this upregulation excludes genes involved in beta-cell function. -
Potential Role of Increased Oxygenation in Altering Perinatal Adrenal Steroidogenesis
nature publishing group Articles Basic Science Investigation Potential role of increased oxygenation in altering perinatal adrenal steroidogenesis Vishal Agrawal1, Meng Kian Tee1, Jie Qiao1, Marcus O. Muench2,3 and Walter L. Miller1 BACKGROUND: At birth, the large fetal adrenal involutes rap- respond to adrenocorticotropic hormone (1). The fetal adrenal idly, and the patterns of steroidogenesis change dramatically; transiently expresses 3β-hydroxysteroid dehydrogenase, type the event(s) triggering these changes remain largely unex- 2 (3βHSD2, encoded by HSD3B2) at about 8–10 wk, permit- plored. Fetal abdominal viscera receive hypoxic blood having a ting fetal adrenal cortisol synthesis at the same time when male partial pressure of oxygen of only ~2 kPa (20–23 mm Hg); peri- genital development occurs, thus helping to prevent the viril- natal circulatory changes change this to adult values (~20 kPa). ization of female fetuses by suppressing fetal adrenal andro- We hypothesized that transition from fetal hypoxia to postna- gen synthesis (1). The fetal adrenal has relatively little 3βHSD2 tal normoxia participates in altering perinatal steroidogenesis. activity after 12 wk (1,2) but has 17α-hydroxylase and robust METHODS: We grew midgestation human fetal adrenal cells 17,20 lyase activity (both catalyzed by cytochrome P450c17, and human NCI-H295A adrenocortical carcinoma cells in 2% encoded by CYP17A1), considerable sulfotransferase activity, O2, then transitioned them to 20% O2 and quantitated ste- and little steroid sulfatase activity accounting for its abun- roidogenic mRNAs by quantitative PCR and microarrays. dant production of dehydroepiandrosterone (DHEA) and its RESULTS: Transitioning fetal adrenal cells from hypoxia to sulfate (DHEAS). DHEAS is secreted, 16α-hydroxylated in normoxia increased mRNAs for 17α-hydroxylase/17,20 lyase the fetal liver by CYP3A7 (3–5), and then acted on by pla- (P450c17), 3β-hydroxysteroid dehydrogenase (3βHSD2), and cental 3βHSD1, 17βHSD1 (17β-hydroxysteroid dehydroge- steroidogenic acute regulatory protein (StAR). -

WD40-Repeat Proteins in Ciliopathies and Congenital Disorders of Endocrine System
ARTICLE IN PRESS Review Endocrinol Metab 2020 Forthcoming. https://doi.org/10.3803/EnM.2020.302 Article pISSN 2093-596X · eISSN 2093-5978 WD40-Repeat Proteins in Ciliopathies and Congenital Disorders of Endocrine System Yeonjoo Kim, Soo-Hyun Kim Cell Biology Research Centre, Molecular and Clinical Sciences Research Institute, St. George’s, University of London, London, UK WD40-repeat (WDR)-containing proteins constitute an evolutionarily conserved large protein family with a broad range of biologi- cal functions. In human proteome, WDR makes up one of the most abundant protein-protein interaction domains. Members of the WDR protein family play important roles in nearly all major cellular signalling pathways. Mutations of WDR proteins have been as- sociated with various human pathologies including neurological disorders, cancer, obesity, ciliopathies and endocrine disorders. This review provides an updated overview of the biological functions of WDR proteins and their mutations found in congenital disorders. We also highlight the significant role of WDR proteins in ciliopathies and endocrine disorders. The new insights may help develop therapeutic approaches targeting WDR motifs. Keywords: WDR proteins; Ciliopathies; Congenital, hereditary, and neonatal diseases and abnormalities; Neuroendocrine; Kall- mann syndrome INTRODUCTION proteins (based on UniProt) discussed in this review are sum- marised in the Supplemental Table S1. WDR proteins associated WD40-repeat (WDR) refers to a series of loosely conserved with pathological conditions that are not discussed in the main structural motifs comprised of approximately 40 amino acids, text in detail are summarised in the Supplemental Table S2. often terminating in tryptophan (W)-aspartic acid (D). WDR protein family is a large group of proteins commonly possessing MOLECULAR STRUCTURE OF WDR the WDR motifs, that are involved in a wide range of important PROTEINS biological processes. -
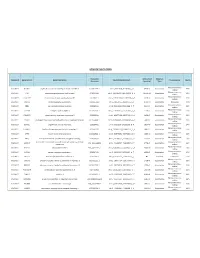
Somatic Mutations
SOMATIC MUTATIONS Transcript Amino Acid Mutation Sample ID Gene Symbol Gene Description Nucleotide (genomic) Consequence Mut % Accession (protein) Type Nonsynonymous PGDX11T ACSBG2 acyl-CoA synthetase bubblegum family member 2 CCDS12159.1 chr19_6141628_6141628_C_A 654A>D Substitution 46% coding Nonsynonymous PGDX11T ATR ataxia telangiectasia and Rad3 related CCDS3124.1 chr3_143721232_143721232_G_A 1451R>W Substitution 32% coding Nonsynonymous PGDX11T C1orf183 chromosome 1 open reading frame 183 CCDS841.1 chr1_112071336_112071336_C_T 224R>Q Substitution 22% coding PGDX11T CMYA5 cardiomyopathy associated 5 NM_153610 chr5_79063455_79063455_C_A 1037C>X Substitution Nonsense 34% Nonsynonymous PGDX11T CNR1 cannabinoid receptor 1 (brain) CCDS5015.1 chr6_88911646_88911646_C_T 23V>M Substitution 35% coding Nonsynonymous PGDX11T COL4A4 collagen; type IV; alpha 4 CCDS42828.1 chr2_227681807_227681807_G_A 227R>C Substitution 20% coding Nonsynonymous PGDX11T CYBASC3 cytochrome b; ascorbate dependent 3 CCDS8004.1 chr11_60877136_60877136_G_A 149R>C Substitution 34% coding Nonsynonymous PGDX11T DYRK3 dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 3 CCDS30999.1 chr1_204888091_204888091_G_A 309V>I Substitution 44% coding Nonsynonymous PGDX11T ELMO1 engulfment and cell motility 1 CCDS5449.1 chr7_37239295_37239295_G_A 160T>M Substitution 24% coding Nonsynonymous PGDX11T FAM83H family with sequence similarity 83; member H CCDS6410.2 chr8_144884473_144884473_C_A 90G>C Substitution 53% coding Nonsynonymous PGDX11T KPTN kaptin (actin binding protein) -
Supplementary Table 1 Gene Primer Sequence S6K1 F Taaagggggctatggaaagg S6K1 R Ttaagcaccttcatggcaaat RASSF6 F Cccaggattttgctcttca
1 Supplementary Table 1 Gene primer sequence S6K1_F taaagggggctatggaaagg S6K1_R ttaagcaccttcatggcaaat RASSF6_F cccaggattttgctcttcac RASSF6_R cctctgcagtagcggaatgt CDKNIA_F ccgaagtcagttccttgtgg CDKNIA_R catgggttctgacggacat DAPK2_F acgtggtgctcatccttga DAPK2_R tggcctcctcctcactca TP53I3_F tctctgaagcaacgctgaaa TP53I3_R gtaggatccgcctatgcagt SKP2_F ctgtctcaaggggtgattgc SKP2_R ttcgataggtccatgtgctg SERBP1_F gcagcaggaccgacaagt SERBP1_R tgtcttatggcatccagttaagc CEACAM1_F cccatcatgctgaacgtaaa CEACAM1_R agggccactactccaatcac GAPDH_F ccccggtttctataaattgagc GAPDH_R cgaacaggaggagcagagag 2 Supplementary Table 2 Clinicopathological Frequency S6K1 overexpression p value* features (%) Mean ± SD/Number of cases Negative Positive Age (year) <59 32 (46.4%) 21 11 0.508 ≥59 37 (53.6%) 27 10 Sex Male 51 (73.9%) 38 13 0.133 Female 18 (26.1%) 10 8 Family History Yes 12 (17.4%) 8 4 0.81 No 57 (82.6%) 40 17 Smoke No 37 (53.6%) 22 15 0.05 Yes 32 (46.4%) 26 6 Drink No 47 (68.1%) 31 16 0.341 Yes 22 (31.9%) 17 5 HBV carrier No 16 (23.2%) 11 5 0.936 Yes 53 (76.8%) 37 16 Albumin (g/l) 39.0±4.5 39.1±5.5 0.942 HBsAg Positive 61 (88.4%) 42 19 0.722 Negative 8 (11.6%) 6 2 Tumor recurrence Absence 20 (29.0%) 14 6 0.96 Presence 49 (71.0%) 34 15 Tumor size (cm) 8.0±4.9 8.8±6.1 0.599 Number of tumor nodules 1 46 (66.7%) 31 15 0.579 ≥2 23 (33.3%) 17 6 Differentiation** Well 9 (13.4%) 4 5 0.092 Moderately 42 (70.5%) 29 13 Poorly 15 (16.1%) 13 2 Venous infiltration** Absence 34 (51.5%) 20 14 0.092 Presence 32 (48.5%) 25 7 TNM I/II 31 (44.9%) 19 12 0.177 III/IV 38 (55.1%) 29 9 AJCC I/II -

00117-2019.Tablet3
row.names GeneSym r pval n IPFsig Description ENSG00000197818 SLC9A8 0.628861777 5.21123E-05 35 FALSE solute carrier family 9 member A8 ENSG00000163428 LRRC58 -0.628581035 5.26373E-05 35 FALSE leucine rich repeat containing 58 ENSG00000070214 SLC44A1 -0.62479102 6.02081E-05 35 TRUE solute carrier family 44 member 1 ENSG00000171951 SCG2 -0.588154207 0.000202637 35 TRUE secretogranin II ENSG00000166454 ATMIN -0.616509135 8.02719E-05 35 FALSE ATM interactor ENSG00000122591 FAM126A -0.614824684 8.50235E-05 35 FALSE family with sequence similarity 126 member A ENSG00000112200 ZNF451 -0.610753927 9.75683E-05 35 FALSE zinc finger protein 451 ENSG00000166181 API5 -0.609209846 0.000102746 35 FALSE apoptosis inhibitor 5 ENSG00000127948 POR 0.607665766 0.00010817 35 FALSE cytochrome p450 oxidoreductase ENSG00000178904 DPY19L3 -0.602752784 0.000127181 35 FALSE dpy-19 like 3 (C. elegans) ENSG00000111596 CNOT2 -0.601629816 0.000131927 35 FALSE CCR4-NOT transcription complex subunit 2 ENSG00000004897 CDC27 -0.600226107 0.000138083 35 FALSE cell division cycle 27 ENSG00000169490 TM2D2 -0.59166348 0.000181541 35 FALSE TM2 domain containing 2 ENSG00000110367 DDX6 -0.591242367 0.000183965 35 FALSE DEAD-box helicase 6 ENSG00000124767 GLO1 -0.532567317 0.000989844 35 TRUE glyoxalase I ENSG00000143195 ILDR2 -0.580714547 0.000254767 35 FALSE immunoglobulin like domain containing receptor 2 ENSG00000119812 FAM98A -0.577626387 0.000279714 35 FALSE family with sequence similarity 98 member A ENSG00000145390 USP53 -0.528075448 0.001112177 35 TRUE ubiquitin specific -

Examination of Kinase-Regulated Transcriptional Networks
Examinationof Kinase-RegulatedTranscriptional Networks Using Pharmacolosicaland ChemicalGenetic Tools by BeatriceT. Wanq DISSERTATION Submittedin partialsatisfaction of the requirementsfor the degreeof DOCTOROF PHILOSOPHY in Biochemistryand Molecular Biology in the GRADUATE DIVISION of the Copyright 2011 by Beatrice T. Wang ii Acknowledgements First and foremost, I thank my academic advisor Dr. Kevan Shokat for his guidance and support throughout my graduate career. Kevan has a gift for understanding complex biological systems and I was so fortunate to have had the opportunity to study under his mentorship. Even during difficult times, Kevan’s insight and perpetual optimism gave me the motivation I needed to succeed, and his admirable ability to achieve a work-life balance makes him an inspiring role model. I was also fortunate to have had the most kind and talented group of colleagues in the Shokat lab. In particular, I am indebted to Jasmina Allen for her dedicated mentorship that was crucial to my graduate learning experience, and to Morri Feldman for always generously offering me his time and ideas. Additionally, I appreciate Sasha Statsuk, Greg Ducker, Adam Garske, Ulf Peters, and Brandon Tavshanjian for not only technical advice but also insightful and entertaining conversations. I also thank Valerie Ohman for providing impeccable administrative assistance that the lab could not function without. I am also grateful to my thesis committee, Keith Yamamoto and Geeta Narlikar, for providing scientific support and career advice. I was supported in 2008-2009 by the University of California Cancer Research Coordinating Committee. Lastly, I would like to acknowledge my family, Ping, Teresa, and Clarice Wang for their love and support. -
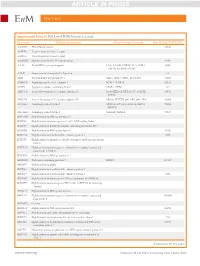
Article in Press
ARTICLE IN PRESS Kim Y, et al. Supplemental Table S1. Full List of WDR Proteins Accessed Gene name Full name of the protein Alternative name of the gene Gene ID from NCBI RefSeq A6NM71 PRA1 family protein 10567 A6P4T4 Tyrosine-protein kinase receptor A6P4V4 Tyrosine-protein kinase receptor A8MWR8 Highly similar to SEC13-related protein 6396 AAAS Aladin WD repeat nucleoporin AAA, AAASb, ADRACALA, ADRA- 8086 CALIN, ALADIN, GL003 AAMP Angio associated migratory cell protein 14 AHI1 Abelson helper integration site 1 AHI-1, JBTS3, ORF1, dJ71N10.1 54806 AMBRA1 Autophagy and beclin 1 regulator 1 DCAF3, WDR94 55626 APAF1 Apoptotic peptidase activating factor 1 APAF-1, CED4 317 ARPC1A Actin related protein 2/3 complex subunit 1A Arc40, HEL-68, HEL-S-307, SOP2Hs, 10552 SOP2L ARPC1B Actin related protein 2/3 complex subunit 1B ARC41, PLTEID, p40-ARC, p41-ARC 10095 ATG16L1 Autophagy related 16 like 1 APG16L, ATG16A, ATG16L, IBD10, 55054 WDR30 ATG16L2 Autophagy related 16 like 2 ATG16B, WDR80 89849 B3KMW5 Highly similar to WD repeat protein 3 B3KP68 Highly similar to homo sapiens selective LIM binding factor B3KP80 Highly similar to BTB/POZ domain-containing protein KCTD3 B3KRR8 Highly similar to WD repeat protein 6 11180 B3KUA2 Highly similar to transducin-like enhancer protein 3 7090 B3KVI6 Highly similar to lipopolysaccharide-responsive and beige-like anchor protein B3KXA3 Highly similar to homo sapiens echinoderm microtubule associated protein like 1 (EML1) B3KXN4 Highly similar to WD repeat protein 1 B4DDD4 WD repeat-containing protein 27 WDR27