Origin and Fate of Supergenes in Atlantic Cod
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Complete Mitochondrial Genome Sequences of the Arctic Ocean Codwshes Arctogadus Glacialis and Boreogadus Saida Reveal Oril and Trna Gene Duplications
Polar Biol (2008) 31:1245–1252 DOI 10.1007/s00300-008-0463-7 ORIGINAL PAPER Complete mitochondrial genome sequences of the Arctic Ocean codWshes Arctogadus glacialis and Boreogadus saida reveal oriL and tRNA gene duplications Ragna Breines · Anita Ursvik · Marianne Nymark · Steinar D. Johansen · Dag H. Coucheron Received: 4 December 2007 / Revised: 16 April 2008 / Accepted: 5 May 2008 / Published online: 27 May 2008 © The Author(s) 2008 Abstract We have determined the complete mitochon- Introduction drial genome sequences of the codWshes Arctogadus gla- cialis and Boreogadus saida (Order Gadiformes, Family More than 375 complete sequenced mitochondrial genomes Gadidae). The 16,644 bp and 16,745 bp mtDNAs, respec- from ray-Wnned Wshes have so far (December 2007) been tively, contain the same set of 37 structural genes found in submitted to the database (http://www.ncbi.nlm.nih.gov), all vertebrates analyzed so far. The gene organization is and many of these sequences have contributed considerably conserved compared to other Gadidae species, but with one to resolving phylogenetic relationships among Wshes. Evo- notable exception. B. saida contains heteroplasmic rear- lutionary relationships at diVerent taxonomic levels have rangement-mediated duplications that include the origin of been addressed, including Division (Inoue et al. 2003; Miya light-strand replication and nearby tRNA genes. Examina- et al. 2003), Subdivision (Ishiguro et al. 2003), Genus tion of the mitochondrial control region of A. glacialis, (Doiron et al. 2002; Minegishi et al. 2005), and Species B. saida, and four additional representative Gadidae genera (Yanagimoto et al. 2004; Ursvik et al. 2007). identiWed a highly variable domain containing tandem The circular mitochondrial genomes from ray-Wnned repeat motifs in A. -

Biodiversity of Arctic Marine Fishes: Taxonomy and Zoogeography
Mar Biodiv DOI 10.1007/s12526-010-0070-z ARCTIC OCEAN DIVERSITY SYNTHESIS Biodiversity of arctic marine fishes: taxonomy and zoogeography Catherine W. Mecklenburg & Peter Rask Møller & Dirk Steinke Received: 3 June 2010 /Revised: 23 September 2010 /Accepted: 1 November 2010 # Senckenberg, Gesellschaft für Naturforschung and Springer 2010 Abstract Taxonomic and distributional information on each Six families in Cottoidei with 72 species and five in fish species found in arctic marine waters is reviewed, and a Zoarcoidei with 55 species account for more than half list of families and species with commentary on distributional (52.5%) the species. This study produced CO1 sequences for records is presented. The list incorporates results from 106 of the 242 species. Sequence variability in the barcode examination of museum collections of arctic marine fishes region permits discrimination of all species. The average dating back to the 1830s. It also incorporates results from sequence variation within species was 0.3% (range 0–3.5%), DNA barcoding, used to complement morphological charac- while the average genetic distance between congeners was ters in evaluating problematic taxa and to assist in identifica- 4.7% (range 3.7–13.3%). The CO1 sequences support tion of specimens collected in recent expeditions. Barcoding taxonomic separation of some species, such as Osmerus results are depicted in a neighbor-joining tree of 880 CO1 dentex and O. mordax and Liparis bathyarcticus and L. (cytochrome c oxidase 1 gene) sequences distributed among gibbus; and synonymy of others, like Myoxocephalus 165 species from the arctic region and adjacent waters, and verrucosus in M. scorpius and Gymnelus knipowitschi in discussed in the family reviews. -
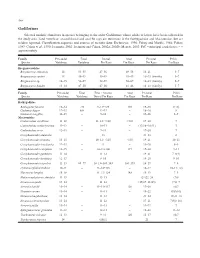
Gadiformes Selected Meristic Characters in Species Belonging to the Order Gadiformes Whose Adults Or Larvae Have Been Collected in the Study Area
548 Gadiformes Selected meristic characters in species belonging to the order Gadiformes whose adults or larvae have been collected in the study area. Total vertebrae, second dorsal and anal fin rays are numerous in the Bathygadidae and Macrouridae, but are seldom reported. Classification sequence and sources of meristic data: Eschmeyer, 1990; Fahay and Markle, 1984; Fahay, 1989; Cohen et al., 1990; Iwamoto, 2002; Iwamoto and Cohen, 2002a; 2002b; Merrett, 2003. PrC = principal caudal rays; ~ = approximately Family Precaudal Total Dorsal Anal Pectoral Pelvic Species Vertebrae Vertebrae Fin Rays Fin Rays Fin Rays Fin Rays Bregmacerotidae Bregmaceros atlanticus 14 53–55 47–56 49–58 16–21 5–7 Bregmaceros cantori 14 45–49 45–49 45–49 16–23 (family) 5–7 Bregmaceros sp. 14–15 52–59 52–59 58–69 16–23 (family) 5–7 Bregmaceros houdei 13–14 47–50 47–50 41–46 16–23 (family) 5–7 Family Precaudal Total First + Second Anal Pectoral Pelvic Species Vertebrae Vertebrae Dorsal Fin Rays Fin Rays Fin Rays Fin Rays Bathygadidae Bathygadus favosus 12–14 ~70 9–11+125 110 15–18 9(10) Gadomus dispar 12–13 80+ 12–13 – 18–20 8 Gadomus longifilis 11–13 – 9–11 – 14–16 8–9 Macrouridae Caelorinchus caribbeus 11–12 – 11–12+>110 >110 17–20 7 Caelorinchus coelorhynchus 11–12 – 10–11 – (17)18–20(21) 7 Caelorinchus occa 12–13 – 9–11 – 17–20 7 Coryphaenoides alateralis – 13 – 21–23 8 Coryphaenoides armatus 13–15 – 10–12+~125 ~135 19–21 10–11 Coryphaenoides brevibarbis 12–13 – 9 – 19–20 8–9 Coryphaenoides carapinus 12–15 – 10–11+100 117 17–20 9–11 Coryphaenoides guentheri -

Descriptive Key to the Otoliths of Gadid Fishes of the Bering, Chukchi, and Beaufort Seas KATHRYN J
ARCTIC VOL. 34, NO. 1 (MARCH 1981), P. 55-59 Descriptive Key to the Otoliths of Gadid Fishes of the Bering, Chukchi, and Beaufort Seas KATHRYN J. FROST’ ABSTRACT. An illustrated key with supplementary descriptive material is presented for six species or species groups of gadid fishes which are of trophic importance in the Bering, Chukchi, and Beaufort seas. These species include: Arcrogadus spp. Djagin, Boreogadus saida (Lepechin), Eleginus gracilis (Tilesius), Gadus macrocephalus Tilesius, Microgadus proximus (Girard), and Theragra chalcogramma (Pallas). RESUME. Une clC d’identificationillustree par des figures avec un complement descriptif est ici presentee pour six espbces ou groupes d’espbces de poissons de la famiile des gadidCs, lesquels ont une importance au point de vue trophique dans les mers de BCring, des Tchouktches et deBeaufort. Ces espbcescomprennent: Arctogadus spp. Djagin, Boreogadus saida (Lepechin), Eleginus gracilis (Tilesius), Gadus macrocephalus Tilesius, Microgadus proximus (Girard), et Theragra chalcogramma (Pallas). Traduit par Jean-Guy Brossard, Laboratoire d’Archeologie de I’Universit6 due Quebec A Montreal. Key words: otolith, gadid fishes, Arctogadus, Boreogadus, Eleginus, Gadus, Microgadus, Theragra INTRODUCTION size or when certain features vary such that an otolith of Investigations of food habits of marine animals almost one species closelyresembles that of other species. Furth- invariably involve analysisof stomach contents. Success- er, keys,are often usedby readers who have little familiar- ful stomach contents analysis usually requires that prey ity with otoliths and limited access to comparative mate- items be recognized by characteristic fragments. In this rial, and who therefore require more detailed descriptive respect the sagittal otoliths of bony fishes are very useful material. -

The Ringed Seal, Phoca Hispida, of the Canadian Western Arctic Jimmy Memorana of Holman, Northwest Territories, with a Day's Catch of Ringed Seals
Canadian Bulletin of Fisheries_and Aquatic-Sciences 216 DFO - Lib ary / MPO - B b otheque 11111 I 1111 11111 11 11 12039511 The Ringed Seal, Phoca hispida. of the Canadian Western Arctic Thomas G. Smith -SW D23 92/3 2 C. / Fisheries Pêches 1+ and Oceans et Océans The Ringed Seal, Phoca hispida, of the Canadian Western Arctic Jimmy Memorana of Holman, Northwest Territories, with a day's catch of ringed seals. Il Canadian Bulletin of Fisheries and Aquatic Sciences 216 The Ringed Seal, Phoca hispida, of the Canadian Western Arctic Thomas G. Smith Department of Fisheries and Oceans Arctic Biological Station 555 St-Pierre Blvd. Sainte-Anne-de-Bellevue, Quebec H9X 3R4 DEPARTMENT OF FISHERIES AND OCEANS Ottawa 1987 III The Canadian Bulletins of Fisheries and Aquatic. Sciences are designed to interpret current knowledge in scientific fields pertinent to Canadian fisheries and aquatic environments. The Canadian Journal of Fisheries and Aquatic Sciences is published in annual volumes of monthly issues. Canadian Special Publications of Fisheries and Aquatic Sciences arc issued periodically. These series are available from authorized bookstore agents and other bookstores, or you may send your prepaid order to the Canadian Government Publishing Centre, Supply and Services Canada, Ottawa, Ont. KIA 0S9. Make cheque or money order payable in Canadian funds to the Receiver General for Canada. Information and Publications Branch Dixi Lambert Director General Johanna M. Reinhart, M.Sc. Director and Editor Gerald J. Neville Editorial and Publishing Services Editorial Office: Department of Fisheries and Oceans Communications Directorate Information and Publications Branch 200 Kent Street Ottawa, Ontario, Canada K IA 0E6 Typesetter: K.G. -

BOEM 2017-066 Genomics of Arctic
OCS Study BOEM 2017 -066 Genomics of Arctic Cod US Department of the Interior Bureau of Ocean Energy Management Alaska OCS Region OCS Study BOEM 2017-066 Genomics of Arctic Cod 30 September 2017 Authors Robert E. Wilson, George K. Sage, Sarah A. Sonsthagen, Meg C. Gravley, Damian M. Menning and Sandra L. Talbot Prepared under BOEM Award M14PG00008 By U. S. Geological Survey, Alaska Science Center 4210 University Drive Anchorage, AK 99508 USA US Department of the Interior Bureau of Ocean Energy Management Alaska OCS Region DISCLAIMER This study was funded, in part, by the US Department of the Interior, Bureau of Ocean Energy Management (BOEM), Environmental Studies Program, Washington, DC, through Agreement Number M14PG00008 with the U.S. Geological Survey (USGS). The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the Bureau of Oceans Energy and Management. REPORT AVAILABILITY To download a PDF file of this report, go to the U.S. Department of the Interior, Bureau of Ocean Energy Management Data and Information Systems webpage (https://marinecadastre.gov/espis/#) and search on 2017-066. CITATION Wilson, R.E, G.K. Sage, S.A. Sonsthagen, M.C. Gravley, D.M. Menning, and S.L. Talbot. 2017. Genomics of Arctic Cod. Anchorage, AK: US Dept. of the Interior, Bureau of Ocean Energy Management, Alaska OCS Region. OCS Study BOEM 2017-066. 92pp. ACKNOWLEDGEMENTS We thank the numerous staff at U.S. Geological Survey for providing samples and for laboratory and graphical assistance. The BOEM Alaska OCS Region contributed to this document. -

Oxygen Consumption of East Siberian Cod: No Support for the Metabolic Cold Adaptation Theory
Journal of Fish Biology (2001) 59, 818–823 doi:10.1006/jfbi.2001.1692, available online at http://www.idealibrary.com on Oxygen consumption of East Siberian cod: no support for the metabolic cold adaptation theory A. D J,M.J J. F. S* The Arctic Station, University of Copenhagen, DK-3953 Godhavn/Qeqertarsuaq, Greenland and The Marine Biological Laboratory, University of Copenhagen, Strandpromenaden 5, DK-3000 Helsingør, Denmark (Received 7 January 2001, Accepted 18 June 2001) Standard metabolic rate (RS)at2 C of eight East Siberian cod Arctogadus borisovi, caught in 1 1 West Greenland, body mass of 601·5 147·6 g (mean . .), was 40·9 5·9 mg O2 kg h 1 1 and 59·0 6·6 mg O2 kg h when extrapolated to a standardized 100 g fish. RS was compared with three other Gadidae, to test the theory of metabolic cold adaptation (MCA). There was no evidence of MCA in the family. 2001 The Fisheries Society of the British Isles Key words: East Siberian cod; Arctogadus borisovi; Arctic; metabolic cold adaptation; oxygen consumption. INTRODUCTION The classical studies of Ege & Krogh (1914) and Krogh (1914, 1916) suggested that polar ectotherms would have an elevated metabolic rate (metabolic cold adaptation, MCA) relative to temperate ectotherms when exposed to the same temperature. Work by Scholander et al. (1953) and Wohlschlag (1960, 1964) suggested that cold-water fishes tended to have a relatively higher metabolic rate than temperate and warm-water species. The theory was later criticized (Holeton, 1973, 1974; Clarke, 1980, 1983, 1991, 1993; Hop & Graham, 1995), and several authors claimed that there was no evidence for MCA (Wells, 1986, 1987; Bushnell et al., 1994; Steffensen et al., 1994; Clarke & Johnston, 1999). -

Walleye Pollock Gadus Chalcogrammus Pallas, 1814 Found North of Spitsbergen Indicates a Pacific-Atlantic Connection in the Species
Fauna norvegica 2020 Vol. 40: 137–140. Short communication Walleye pollock Gadus chalcogrammus Pallas, 1814 found north of Spitsbergen indicates a Pacific-Atlantic connection in the species Ingvar Byrkjedal1 and Gunnar Langhelle1 Byrkjedal I and Langhelle A. 2020. Walleye pollock Gadus chalcogrammus Pallas, 1814 found north of Spitsbergen indicates a Pacific-Atlantic connection in the species. Fauna norvegica 40: 137–140. A small (36.5 cm) female walleye pollock Gadus chalcogrammus Pallas, 1814 was caught north of Spitsbergen in 2011. The specimen had a high number of pyloric caeca (n=109), indicating a Pacific origin. The record is the first evidence of a possible input to the small Atlantic population of walleye pollock from the large populations on the Pacific side. doi: 10.5324/fn.v40i0.3394. Received: 2020-01-21. Accepted: 2020-09-16. Published online: 2020-10-16. ISSN: 1891-5396 (electronic). Keywords: Gadoids, distribution, population exchange 1. Department of Natural History, University Museum of Bergen, University of Bergen, NO-5020 Bergen, Norway Corresponding author: Ingvar Byrkjedal E-mail: [email protected] INTRODUCTION differences between the Atlantic and Pacific populations have been The walleye pollock Gadus chalcogrammus Pallas, 1814 has its found (Byrkjedal et al. 2008). We here report on a specimen caught primary distribution in the North Pacific, where it is abundant and in 2011 north of Spitsbergen and discuss the possible origin of this of prime commercial value (Mecklenburg et al. 2018). A population specimen from examination of population-distinguishing characters. also exists in the Northeast Atlantic, found mostly in the Barents Sea, from the coast of North Norway to as far north as near 76oN and east to 42o38’E. -

Ice Cod to Pacific
Ice Cod 183 Ice Cod to Pacific Cod Ice Cod (Arctogadus glacialis) (Peters, 1872) Family Gadidae Note on taxonomy: Evidence from morphology and molecular genetics demonstrates that Arctogadus borisovi (Dryagin, 1932) is a junior synonym of A. glacialis [1]. Data on fish originally identified as A. borisovi are included here. Commmonly referred to Ice Cod (Arctogadus glacialis) 221 mm, Chukchi Borderland, as Polar Cod in North America. 2009. Photograph by C.W. Mecklenburg, Point Stephens Colloquial Name: None within U.S. Chukchi and Beaufort Seas. Research. Ecological Role: The ecological role of the species in marine ecosystems of the U.S. Chukchi and Beaufort Seas is not as significant as Polar and Saffron Cod. Physical Description/Attributes: An olive brown to bluish gray cod with darker fins and head. For specific diagnostic characteristics, see Fishes of Alaska (Mecklenburg and others, 2002, p. 291–292) [2]. Swim bladder: Present; no otophysic connection [2]. Antifreeze glycoproteins in blood serum: Unknown. Range: U.S. Beaufort [2] and Chukchi Sea [3, 4]. Worldwide, circumpolar, northward to at least 81°41’N; Arctic Canada south to southern tip of Greenland, east through Barents Sea to East Siberian Sea and Chukchi Sea [2–4]. 184 Alaska Arctic Marine Fish Ecology Catalog Relative Abundance: Rare in U.S. Beaufort Sea (two specimens captured north of Point Barrow) [2] and Chukchi Sea (one specimen found on beach at Wainwright) [4].Abundant to at least as far eastward to deep waters off Tuktoyaktuk Peninsula and off Capes Bathurst and -

A Review of the Impact of Seismic Survey Noise on Narwhal and Other
A Review of the Impact of Seismic Survey Noise on Narwhal & other Arctic Cetaceans Report prepared for Greenpeace Nordic by Marine Conservation Research Ltd. Prepared by Cucknell, A-C., Boisseau, O., Moscrop, A. May 2015 1 Contents Executive Summary ................................................................................................................................ 7 Glossary of terms relating to seismic testing ........................................................................................ 10 1. Introduction .................................................................................................................................. 12 2. Seismic testing that will be conducted in Canadian waters of Baffin Bay in 2015 ....................... 16 Summary ....................................................................................................................................... 16 2.1 General background and information .................................................................................. 16 2.2 Brief description of the seismic program .............................................................................. 17 2.3 Seismic data acquisition: ....................................................................................................... 18 2.4 Seismic and support vessel operations: ................................................................................ 19 2.5 NEB’s Environmental Assessment Report ............................................................................ -

Latitudinal Cline in Chromosome Numbers of Ice Cod A
G C A T T A C G G C A T genes Article Latitudinal Cline in Chromosome Numbers of Ice Cod A. glacialis (Gadidae) from Northeast Greenland Laura Ghigliotti 1,* , Jørgen S. Christiansen 2,3, Erica Carlig 1, Davide Di Blasi 1 and Eva Pisano 1 1 National Research Council of Italy, Institute for the Study of the Anthropic Impacts and the Sustainability of the Marine Environment (IAS) via De Marini 6, 16149 Genoa, Italy; [email protected] (E.C.); [email protected] (D.D.B.); [email protected] (E.P.) 2 Department of Arctic and Marine Biology, UIT The Arctic University of Norway, NO-9037 Tromsø, Norway; [email protected] 3 Department of Environmental and Marine Biology, Åbo Akademi University, FI-20520 Turku, Finland * Correspondence: [email protected] or [email protected] Received: 20 November 2020; Accepted: 15 December 2020; Published: 18 December 2020 Abstract: The ice cod Arctogadus glacialis (Peters, 1872) is one of the few fish species endemic to the Arctic. With a circumpolar distribution, the species is confined to the fjords and shelves of the Arctic seas. Biological information on A. glacialis is scarce, with genomic information restricted to microsatellites. Within the frame of the TUNU-Programme: Arctic Ocean Fishes—Diversity, Adaptation and Conservation, we studied A. glacialis at the chromosomal level to explore fish diversity and evolutionary aspects. The analysis of over 50 individuals from the Northeast Greenland fjords between latitudes 71◦090 N and 76◦420 N revealed a remarkable intraspecific diversity epitomized by chromosome numbers spanning from 28 to 33, the occurrence of putative B chromosomes, and diversified patterns of distribution of heterochromatin and rDNAs. -

Phoca Hispida)
Background Document for Development of a Circumpolar Ringed Seal (Phoca hispida) Monitoring Plan Photo: Kit M. Kovacs & Christian Lydersen Prepared by Kit M. Kovacs Table of Contents I. Ecology of the species.................................................................................................. 3 A. Brief review of biology and natural history....................................................... 3 1. Basic biology.................................................................................................... 3 2. General distribution and abundance patterns ............................................. 4 3. Reproductive biology...................................................................................... 6 4. Movements/migrations ................................................................................... 9 5. Seasonal/regional variation.......................................................................... 10 6. Foraging......................................................................................................... 11 7. Competition for prey .................................................................................... 13 8. Predators........................................................................................................ 13 9. Human harvests ............................................................................................ 14 10. Health (disease, parasites, contaminants)................................................. 15 B. Climate change in the Arctic and its