Multiple Gene Mutations Identified in Patients Infected with Influenza A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information Integrative Analyses of Splicing in the Aging Brain: Role in Susceptibility to Alzheimer’S Disease
Supplementary Information Integrative analyses of splicing in the aging brain: role in susceptibility to Alzheimer’s Disease Contents 1. Supplementary Notes 1.1. Religious Orders Study and Memory and Aging Project 1.2. Mount Sinai Brain Bank Alzheimer’s Disease 1.3. CommonMind Consortium 1.4. Data Availability 2. Supplementary Tables 3. Supplementary Figures Note: Supplementary Tables are provided as separate Excel files. 1. Supplementary Notes 1.1. Religious Orders Study and Memory and Aging Project Gene expression data1. Gene expression data were generated using RNA- sequencing from Dorsolateral Prefrontal Cortex (DLPFC) of 540 individuals, at an average sequence depth of 90M reads. Detailed description of data generation and processing was previously described2 (Mostafavi, Gaiteri et al., under review). Samples were submitted to the Broad Institute’s Genomics Platform for transcriptome analysis following the dUTP protocol with Poly(A) selection developed by Levin and colleagues3. All samples were chosen to pass two initial quality filters: RNA integrity (RIN) score >5 and quantity threshold of 5 ug (and were selected from a larger set of 724 samples). Sequencing was performed on the Illumina HiSeq with 101bp paired-end reads and achieved coverage of 150M reads of the first 12 samples. These 12 samples will serve as a deep coverage reference and included 2 males and 2 females of nonimpaired, mild cognitive impaired, and Alzheimer's cases. The remaining samples were sequenced with target coverage of 50M reads; the mean coverage for the samples passing QC is 95 million reads (median 90 million reads). The libraries were constructed and pooled according to the RIN scores such that similar RIN scores would be pooled together. -

Dual Proteome-Scale Networks Reveal Cell-Specific Remodeling of the Human Interactome
bioRxiv preprint doi: https://doi.org/10.1101/2020.01.19.905109; this version posted January 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Dual Proteome-scale Networks Reveal Cell-specific Remodeling of the Human Interactome Edward L. Huttlin1*, Raphael J. Bruckner1,3, Jose Navarrete-Perea1, Joe R. Cannon1,4, Kurt Baltier1,5, Fana Gebreab1, Melanie P. Gygi1, Alexandra Thornock1, Gabriela Zarraga1,6, Stanley Tam1,7, John Szpyt1, Alexandra Panov1, Hannah Parzen1,8, Sipei Fu1, Arvene Golbazi1, Eila Maenpaa1, Keegan Stricker1, Sanjukta Guha Thakurta1, Ramin Rad1, Joshua Pan2, David P. Nusinow1, Joao A. Paulo1, Devin K. Schweppe1, Laura Pontano Vaites1, J. Wade Harper1*, Steven P. Gygi1*# 1Department of Cell Biology, Harvard Medical School, Boston, MA, 02115, USA. 2Broad Institute, Cambridge, MA, 02142, USA. 3Present address: ICCB-Longwood Screening Facility, Harvard Medical School, Boston, MA, 02115, USA. 4Present address: Merck, West Point, PA, 19486, USA. 5Present address: IQ Proteomics, Cambridge, MA, 02139, USA. 6Present address: Vor Biopharma, Cambridge, MA, 02142, USA. 7Present address: Rubius Therapeutics, Cambridge, MA, 02139, USA. 8Present address: RPS North America, South Kingstown, RI, 02879, USA. *Correspondence: [email protected] (E.L.H.), [email protected] (J.W.H.), [email protected] (S.P.G.) #Lead Contact: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2020.01.19.905109; this version posted January 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. -

Distinguishing Pleiotropy from Linked QTL Between Milk Production Traits
Cai et al. Genet Sel Evol (2020) 52:19 https://doi.org/10.1186/s12711-020-00538-6 Genetics Selection Evolution RESEARCH ARTICLE Open Access Distinguishing pleiotropy from linked QTL between milk production traits and mastitis resistance in Nordic Holstein cattle Zexi Cai1*†, Magdalena Dusza2†, Bernt Guldbrandtsen1, Mogens Sandø Lund1 and Goutam Sahana1 Abstract Background: Production and health traits are central in cattle breeding. Advances in next-generation sequencing technologies and genotype imputation have increased the resolution of gene mapping based on genome-wide association studies (GWAS). Thus, numerous candidate genes that afect milk yield, milk composition, and mastitis resistance in dairy cattle are reported in the literature. Efect-bearing variants often afect multiple traits. Because the detection of overlapping quantitative trait loci (QTL) regions from single-trait GWAS is too inaccurate and subjective, multi-trait analysis is a better approach to detect pleiotropic efects of variants in candidate genes. However, large sample sizes are required to achieve sufcient power. Multi-trait meta-analysis is one approach to deal with this prob- lem. Thus, we performed two multi-trait meta-analyses, one for three milk production traits (milk yield, protein yield and fat yield), and one for milk yield and mastitis resistance. Results: For highly correlated traits, the power to detect pleiotropy was increased by multi-trait meta-analysis com- pared with the subjective assessment of overlapping of single-trait QTL confdence intervals. Pleiotropic efects of lead single nucleotide polymorphisms (SNPs) that were detected from the multi-trait meta-analysis were confrmed by bivariate association analysis. The previously reported pleiotropic efects of variants within the DGAT1 and MGST1 genes on three milk production traits, and pleiotropic efects of variants in GHR on milk yield and fat yield were con- frmed. -
Single-Cell Profiling Identifies Impaired Adaptive NK Cells Expanded After HCMV Reactivation in Haploidentical HSCT
Single-cell profiling identifies impaired adaptive NK cells expanded after HCMV reactivation in haploidentical HSCT Elisa Zaghi, … , Enrico Lugli, Domenico Mavilio JCI Insight. 2021;6(12):e146973. https://doi.org/10.1172/jci.insight.146973. Research Article Hematology Immunology Graphical abstract Find the latest version: https://jci.me/146973/pdf RESEARCH ARTICLE Single-cell profiling identifies impaired adaptive NK cells expanded after HCMV reactivation in haploidentical HSCT Elisa Zaghi,1 Michela Calvi,1,2 Simone Puccio,3 Gianmarco Spata,1 Sara Terzoli,1 Clelia Peano,4 Alessandra Roberto,3 Federica De Paoli,3 Jasper J.P. van Beek,3 Jacopo Mariotti,5 Chiara De Philippis,5 Barbara Sarina,5 Rossana Mineri,6 Stefania Bramanti,5 Armando Santoro,5 Vu Thuy Khanh Le-Trilling,7 Mirko Trilling,7 Emanuela Marcenaro,8 Luca Castagna,5 Clara Di Vito,1,2 Enrico Lugli,3,9 and Domenico Mavilio1,2 1Unit of Clinical and Experimental Immunology, IRCCS Humanitas Research Hospital, Rozzano, Milan, Italy. 2BIOMETRA, Università degli Studi di Milano, Milan, Italy. 3Laboratory of Translational Immunology, 4Institute of Genetic and Biomedical Research, UoS Milan, National Research Council, and Genomic Unit, 5Bone Marrow Transplant Unit, and 6Molecular Biology Section, Clinical Investigation Laboratory, IRCCS Humanitas Research Hospital, Milan, Italy. 7Institute for Virology, University Hospital Essen, University Duisburg-Essen, Essen, Germany. 8Department of Experimental Medicine, University of Genoa, Genoa, Italy. 9Flow Cytometry Core, IRCCS Humanitas Research Hospital, Milan, Italy. Haploidentical hematopoietic stem cell transplantation (h-HSCT) represents an efficient curative approach for patients affected by hematologic malignancies in which the reduced intensity conditioning induces a state of immunologic tolerance between donor and recipient. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Supporting Info
advances.sciencemag.org/cgi/content/full/6/29/eaba9589/DC1 Supplementary Materials for Cell invasion in digital microfluidic microgel systems Bingyu B. Li, Erica Y. Scott, M. Dean Chamberlain, Bill T. V. Duong, Shuailong Zhang, Susan J. Done, Aaron R. Wheeler* *Corresponding author. Email: [email protected] Published 15 July 2020, Sci. Adv. 6, eaba9589 (2020) DOI: 10.1126/sciadv.aba9589 This PDF file includes: Figs. S1 to S8 Tables S1 and S2 References Supplementary Figure S1 CIMMS microgels and cell viability. For (a-c) core-shell microgels were formed from 2.4 mg/mL collagen I and 10% BM extract. (a) Cartoon indicating the orientation of the microgel shown in the images. SEM images of (b) the collagen I core (scale bar = 20 µm) and (c) the edge of microgel with core on the "top" and the shell on the "bottom" (scale bar = 100 µm). The pore size of the collagen I core was calculated to be 3.1 m by the method reported by Blackmon et al. (53), which is nearly identical to the pore size reported for the same concentration of collagen I cast in well plates by Lang et al. (54). For (d-f) MDA-MB-231 cells were seeded onto simple collagen I microgels (2.4 mg/mL). (d) Cell count per droplet with seeding density at 200,000 (lt. pink), 400,000 (fuscia) or 600,000 cells/mL (burgundy). Error bars represent ± 1 std. dev. from n = 4 microgels per condition. (e) 3D confocal fluorescent microscopy image of cells stained on day 4 with calcein-AM (live-green) and propidium iodide (dead-red). -

High-Dimensional Analysis Reveals a Distinct Population of Adaptive-Like Tissue
bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.883785; this version posted December 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 High-dimensional analysis reveals a distinct population of adaptive-like tissue- 2 resident NK cells in human lung 3 4 Nicole Marquardt1*, Marlena Scharenberg1, Jeffrey E. Mold2, Joanna Hård2, Eliisa 5 Kekäläinen3,4, Marcus Buggert1, Son Nguyen5,6, Jennifer N. Wilson1, Mamdoh Al- 6 Ameri7, Hans-Gustaf Ljunggren1, Jakob Michaëlsson1 7 8 Running title: Adaptive-like human lung tissue-resident NK cells 9 10 Affiliations: 11 1Center for Infectious Medicine, Department of Medicine, Karolinska Institutet, 12 Stockholm, Sweden 13 2Department of Cell and Molecular Biology, Karolinska Institutet, Stockholm, Sweden 14 3Translational Immunology Research Program & Department of Bacteriology and 15 Immunology, University of Helsinki, Finland 16 4HUSLAB, Division of Clinical Microbiology, Helsinki University Hospital, Helsinki, 17 Finland, 18 5Department of Microbiology, Perelman School of Medicine, University of 19 Pennsylvania, Philadelphia, PA, USA 20 6Institute for Immunology, Perelman School of Medicine, University of Pennsylvania, 21 Philadelphia, PA, USA 22 7Thoracic Surgery, Department of Molecular Medicine and Surgery, Karolinska 23 Institutet, Karolinska University Hospital, Stockholm, Sweden 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2019.12.20.883785; this version posted December 20, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
Type of the Paper (Article
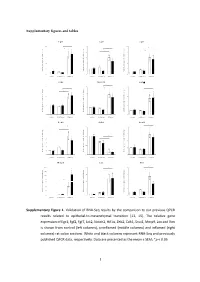
Supplementary figures and tables E g r 1 F g f2 F g f7 1 0 * 5 1 0 * * e e e * g g g * n n n * a a a 8 4 * 8 h h h * c c c d d d * l l l o o o * f f f * n n n o o o 6 3 6 i i i s s s s s s e e e r r r p p p x x x e e e 4 2 4 e e e n n n e e e g g g e e e v v v i i i t t t 2 1 2 a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d J a k 2 N o tc h 2 H if1 * 3 4 6 * * * e e e g g g n n n a a * * a * h h * h c c c 3 * d d * d l l l * o o o f f 2 f 4 n n n o o o i i i s s s s s s e e e r r 2 r p p p x x x e e e e e e n n n e e 1 e 2 g g g e e 1 e v v v i i i t t t a a a l l l e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Z e b 2 C d h 1 S n a i1 * * 7 1 .5 4 * * e e e g g g 6 n n n * a a a * h h h c c c 3 * d d d l l l 5 o o o f f f 1 .0 * n n n * o o o i i i 4 * s s s s s s e e e r r r 2 p p p x x x 3 e e e e e e n n n e e e 0 .5 g g g 2 e e e 1 v v v i i i t t t a a a * l l l e e e 1 * R R R 0 0 .0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d M m p 9 L o x V im 2 0 0 2 0 8 * * * e e e * g g g 1 5 0 * n n n * a a a * h h h * c c c 1 5 * 6 d d d l l l 1 0 0 o o o f f f n n n o o o i i i 5 0 s s s s s s * e e e r r r 1 0 4 3 0 p p p * x x x e e e * e e e n n n e e e 2 0 g g g e e e 5 2 v v v i i i t t t a a a l l l 1 0 e e e R R R 0 0 0 c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d c o n tro l u n in fla m e d in fla m e d Supplementary Figure 1. -

A High Throughput, Functional Screen of Human Body Mass Index GWAS Loci Using Tissue-Specific Rnai Drosophila Melanogaster Crosses Thomas J
Washington University School of Medicine Digital Commons@Becker Open Access Publications 2018 A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses Thomas J. Baranski Washington University School of Medicine in St. Louis Aldi T. Kraja Washington University School of Medicine in St. Louis Jill L. Fink Washington University School of Medicine in St. Louis Mary Feitosa Washington University School of Medicine in St. Louis Petra A. Lenzini Washington University School of Medicine in St. Louis See next page for additional authors Follow this and additional works at: https://digitalcommons.wustl.edu/open_access_pubs Recommended Citation Baranski, Thomas J.; Kraja, Aldi T.; Fink, Jill L.; Feitosa, Mary; Lenzini, Petra A.; Borecki, Ingrid B.; Liu, Ching-Ti; Cupples, L. Adrienne; North, Kari E.; and Province, Michael A., ,"A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses." PLoS Genetics.14,4. e1007222. (2018). https://digitalcommons.wustl.edu/open_access_pubs/6820 This Open Access Publication is brought to you for free and open access by Digital Commons@Becker. It has been accepted for inclusion in Open Access Publications by an authorized administrator of Digital Commons@Becker. For more information, please contact [email protected]. Authors Thomas J. Baranski, Aldi T. Kraja, Jill L. Fink, Mary Feitosa, Petra A. Lenzini, Ingrid B. Borecki, Ching-Ti Liu, L. Adrienne Cupples, Kari E. North, and Michael A. Province This open access publication is available at Digital Commons@Becker: https://digitalcommons.wustl.edu/open_access_pubs/6820 RESEARCH ARTICLE A high throughput, functional screen of human Body Mass Index GWAS loci using tissue-specific RNAi Drosophila melanogaster crosses Thomas J. -

Potential for Natural Killer Cell-Mediated Antibody-Dependent Cellular Cytotoxicity for Control of Human Cytomegalovirus
Antibodies 2013, 2, 617-635; doi:10.3390/antib2040617 OPEN ACCESS antibodies ISSN 2073-4468 www.mdpi.com/journal/antibodies Review Potential for Natural Killer Cell-Mediated Antibody-Dependent Cellular Cytotoxicity for Control of Human Cytomegalovirus Rebecca J. Aicheler *, Eddie C. Y. Wang, Peter Tomasec, Gavin W. G. Wilkinson and Richard J. Stanton Cardiff Institute of Infection and Immunity, Cardiff University, Cardiff, CF14 4XN, UK; E-Mails: [email protected] (E.C.Y.W.); [email protected] (P.T.); [email protected] (G.W.G.W.); [email protected] (R.J.S.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +44-0-292-068-7319. Received: 31 October 2013; in revised form: 15 November 2013 / Accepted: 27 November 2013 / Published: 10 December 2013 Abstract: Human cytomegalovirus (HCMV) is an important pathogen that infects the majority of the population worldwide, yet, currently, there is no licensed vaccine. Despite HCMV encoding at least seven Natural Killer (NK) cell evasion genes, NK cells remain critical for the control of infection in vivo. Classically Antibody-Dependent Cellular Cytotoxicity (ADCC) is mediated by CD16, which is found on the surface of the NK cell in a complex with FcεRI-γ chains and/or CD3ζ chains. Ninety percent of NK cells express the Fc receptor CD16; thus, they have the potential to initiate ADCC. HCMV has a profound effect on the NK cell repertoire, such that up to 10-fold expansions of NKG2C+ cells can be seen in HCMV seropositive individuals. These NKG2C+ cells are reported to be FcεRI-γ deficient and possess variable levels of CD16+, yet have striking ADCC functions. -

Expansions of Adaptive-Like NK Cells with a Tissue-Resident Phenotype in Human Lung and Blood
Expansions of adaptive-like NK cells with a tissue-resident phenotype in human lung and blood Demi Brownliea,1, Marlena Scharenberga,1, Jeff E. Moldb, Joanna Hårdb, Eliisa Kekäläinenc,d,e, Marcus Buggerta, Son Nguyenf,g, Jennifer N. Wilsona, Mamdoh Al-Amerih, Hans-Gustaf Ljunggrena, Nicole Marquardta,2,3, and Jakob Michaëlssona,2 aCenter for Infectious Medicine, Department of Medicine Huddinge, Karolinska Institutet, 14152 Stockholm, Sweden; bDepartment of Cell and Molecular Biology, Karolinska Institutet, 171 77 Stockholm, Sweden; cTranslational Immunology Research Program, University of Helsinki, 00014 Helsinki, Finland; dDepartment of Bacteriology and Immunology, University of Helsinki, 00014 Helsinki, Finland; eHelsinki University Central Hospital Laboratory, Division of Clinical Microbiology, Helsinki University Hospital, 00290 Helsinki, Finland; fDepartment of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; gInstitute for Immunology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA 19104; and hThoracic Surgery, Department of Molecular Medicine and Surgery, Karolinska University Hospital, Karolinska Institutet, 171 76 Stockholm, Sweden Edited by Marco Colonna, Washington University in St. Louis School of Medicine, St. Louis, MO, and approved January 27, 2021 (received for review August 18, 2020) Human adaptive-like “memory” CD56dimCD16+ natural killer (NK) We and others recently identified a subset of tissue-resident − cells in peripheral blood from cytomegalovirus-seropositive indi- CD49a+CD56brightCD16 NK cells in the human lung (14, 15). viduals have been extensively investigated in recent years and are The human lung is a frequent site of infection with viruses such currently explored as a treatment strategy for hematological can- as influenza virus and HCMV, as well as a reservoir for latent cers. -

Differences in Gene Expression Profiles and Carcinogenesis Pathways Involved in Cisplatin Resistance of Four Types of Cancer
596 ONCOLOGY REPORTS 30: 596-614, 2013 Differences in gene expression profiles and carcinogenesis pathways involved in cisplatin resistance of four types of cancer YONG YANG1,2, HUI LI1,2, SHENGCAI HOU1,2, BIN HU1,2, JIE LIU1,3 and JUN WANG1,3 1Beijing Key Laboratory of Respiratory and Pulmonary Circulation, Capital Medical University, Beijing 100069; 2Department of Thoracic Surgery, Beijing Chao-Yang Hospital, Capital Medical University, Beijing 100020; 3Department of Physiology, Capital Medical University, Beijing 100069, P.R. China Received December 23, 2012; Accepted March 4, 2013 DOI: 10.3892/or.2013.2514 Abstract. Cisplatin-based chemotherapy is the standard Introduction therapy used for the treatment of several types of cancer. However, its efficacy is largely limited by the acquired drug Cisplatin is primarily effective through DNA damage and is resistance. To date, little is known about the RNA expression widely used for the treatment of several types of cancer, such changes in cisplatin-resistant cancers. Identification of the as testicular, lung and ovarian cancer. However, the ability RNAs related to cisplatin resistance may provide specific of cancer cells to become resistant to cisplatin remains a insight into cancer therapy. In the present study, expression significant impediment to successful chemotherapy. Although profiling of 7 cancer cell lines was performed using oligo- previous studies have identified numerous mechanisms in nucleotide microarray analysis data obtained from the GEO cisplatin resistance, it remains a major problem that severely database. Bioinformatic analyses such as the Gene Ontology limits the usefulness of this chemotherapeutic agent. Therefore, (GO) and KEGG pathway were used to identify genes and it is crucial to examine more elaborate mechanisms of cisplatin pathways specifically associated with cisplatin resistance.