(12) Patent Application Publication (10) Pub. No.: US 2007/0142328A1 Chapdelaine Et Al
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

WO 2015/072852 Al 21 May 2015 (21.05.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/072852 Al 21 May 2015 (21.05.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 36/84 (2006.01) A61K 31/5513 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 31/045 (2006.01) A61P 31/22 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 31/522 (2006.01) A61K 45/06 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/NL20 14/050780 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 13 November 2014 (13.1 1.2014) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 61/903,430 13 November 2013 (13. 11.2013) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: RJG DEVELOPMENTS B.V. -

WO 2015/072853 Al 21 May 2015 (21.05.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/072853 Al 21 May 2015 (21.05.2015) P O P C T (51) International Patent Classification: (81) Designated States (unless otherwise indicated, for every A61K 45/06 (2006.01) A61K 31/5513 (2006.01) kind of national protection available): AE, AG, AL, AM, A61K 31/045 (2006.01) A61K 31/5517 (2006.01) AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, A61K 31/522 (2006.01) A61P 31/22 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, A61K 31/551 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (21) International Application Number: KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, PCT/NL20 14/050781 MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (22) International Filing Date: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 13 November 2014 (13.1 1.2014) SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (25) Filing Language: English (84) Designated States (unless otherwise indicated, for every (26) Publication Language: English kind of regional protection available): ARIPO (BW, GH, (30) Priority Data: GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 61/903,433 13 November 2013 (13. -

The Organic Chemistry of Drug Synthesis
The Organic Chemistry of Drug Synthesis VOLUME 2 DANIEL LEDNICER Mead Johnson and Company Evansville, Indiana LESTER A. MITSCHER The University of Kansas School of Pharmacy Department of Medicinal Chemistry Lawrence, Kansas A WILEY-INTERSCIENCE PUBLICATION JOHN WILEY AND SONS, New York • Chichester • Brisbane • Toronto Copyright © 1980 by John Wiley & Sons, Inc. All rights reserved. Published simultaneously in Canada. Reproduction or translation of any part of this work beyond that permitted by Sections 107 or 108 of the 1976 United States Copyright Act without the permission of the copyright owner is unlawful. Requests for permission or further information should be addressed to the Permissions Department, John Wiley & Sons, Inc. Library of Congress Cataloging in Publication Data: Lednicer, Daniel, 1929- The organic chemistry of drug synthesis. "A Wiley-lnterscience publication." 1. Chemistry, Medical and pharmaceutical. 2. Drugs. 3. Chemistry, Organic. I. Mitscher, Lester A., joint author. II. Title. RS421 .L423 615M 91 76-28387 ISBN 0-471-04392-3 Printed in the United States of America 10 987654321 It is our pleasure again to dedicate a book to our helpmeets: Beryle and Betty. "Has it ever occurred to you that medicinal chemists are just like compulsive gamblers: the next compound will be the real winner." R. L. Clark at the 16th National Medicinal Chemistry Symposium, June, 1978. vii Preface The reception accorded "Organic Chemistry of Drug Synthesis11 seems to us to indicate widespread interest in the organic chemistry involved in the search for new pharmaceutical agents. We are only too aware of the fact that the book deals with a limited segment of the field; the earlier volume cannot be considered either comprehensive or completely up to date. -

Pharmaceutical Appendix to the Tariff Schedule 2
Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States (2007) (Rev. 2) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names (INN) which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service (CAS) registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. ABACAVIR 136470-78-5 ACIDUM LIDADRONICUM 63132-38-7 ABAFUNGIN 129639-79-8 ACIDUM SALCAPROZICUM 183990-46-7 ABAMECTIN 65195-55-3 ACIDUM SALCLOBUZICUM 387825-03-8 ABANOQUIL 90402-40-7 ACIFRAN 72420-38-3 ABAPERIDONUM 183849-43-6 ACIPIMOX 51037-30-0 ABARELIX 183552-38-7 ACITAZANOLAST 114607-46-4 ABATACEPTUM 332348-12-6 ACITEMATE 101197-99-3 ABCIXIMAB 143653-53-6 ACITRETIN 55079-83-9 ABECARNIL 111841-85-1 ACIVICIN 42228-92-2 ABETIMUSUM 167362-48-3 ACLANTATE 39633-62-0 ABIRATERONE 154229-19-3 ACLARUBICIN 57576-44-0 ABITESARTAN 137882-98-5 ACLATONIUM NAPADISILATE 55077-30-0 ABLUKAST 96566-25-5 ACODAZOLE 79152-85-5 ABRINEURINUM 178535-93-8 ACOLBIFENUM 182167-02-8 ABUNIDAZOLE 91017-58-2 ACONIAZIDE 13410-86-1 ACADESINE 2627-69-2 ACOTIAMIDUM 185106-16-5 ACAMPROSATE 77337-76-9 -

1 441 702 B1
(19) TZZ__Z _T (11) EP 1 441 702 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 31/4045 (2006.01) A61P 25/20 (2006.01) 10.05.2017 Bulletin 2017/19 A61K 9/20 (2006.01) (21) Application number: 02760523.7 (86) International application number: PCT/IL2002/000662 (22) Date of filing: 12.08.2002 (87) International publication number: WO 2003/015690 (27.02.2003 Gazette 2003/09) (54) METHOD FOR TREATING PRIMARY INSOMNIA VERFAHREN ZUR BEHANDLUNG PRIMÄRER INSOMNIA METHODE DE TRAITEMENT DE L’INSOMNIE PRIMAIRE (84) Designated Contracting States: • ROTH T ET AL: "Consensus for the AT BE BG CH CY CZ DE DK EE ES FI FR GB GR pharmacological management of insomnia in th IE IT LI LU MC NL PT SE SK TR enew millennium", INTERNATIONAL JOURNAL Designated Extension States: OF CLINICAL PRACTICE, MEDICON LT LV RO SI INTERNATIONAL, ESHER, GB, vol. 55, no. 1, 1 January 2001 (2001-01-01), page 10PAGES, (30) Priority: 14.08.2001 IL 14490001 XP002990688, ISSN: 1368-5031 • PERLIS M L ET AL: "Psychophysiological (43) Date of publication of application: insomnia: the behavioural model and a 04.08.2004 Bulletin 2004/32 neurocognitive perspective", JOURNAL OF SLEEP RESEARCH, BLACKWELL SCIENTIFIC, (60) Divisional application: OXFORD, GB, vol. 6, 1 January 1997 (1997-01-01), 16172415.8 / 3 103 443 pages 179-188, XP002990686, ISSN: 0962-1105, DOI: DOI:10.1046/J.1365-2869.1997.00045.X (73) Proprietor: NEURIM PHARMACEUTICALS (1991) • SILVA J A C E ET AL: "Special report from a LIMITED symposium held by the WHO and the World Tel Aviv 69710 (IL) Federation of sleep research societies: an overview of insomnias and related disorders - (72) Inventor: ZISAPEL, Nava recognition, epidemiology and rational 69355 Tel Aviv (IL) management", SLEEP, ALLEN PRESS, LAWRENCE, KS,US, vol. -

The Organic Chemistry of Drug Synthesis
THE ORGANIC CHEMISTRY OF DRUG SYNTHESIS VOLUME 3 DANIEL LEDNICER Analytical Bio-Chemistry Laboratories, Inc. Columbia, Missouri LESTER A. MITSCHER The University of Kansas School of Pharmacy Department of Medicinal Chemistry Lawrence, Kansas A WILEY-INTERSCIENCE PUBLICATION JOHN WILEY AND SONS New York • Chlchester • Brisbane * Toronto • Singapore Copyright © 1984 by John Wiley & Sons, Inc. All rights reserved. Published simultaneously in Canada. Reproduction or translation of any part of this work beyond that permitted by Section 107 or 108 of the 1976 United States Copyright Act without the permission of the copyright owner is unlawful. Requests for permission or further information should be addressed to the Permissions Department, John Wiley & Sons, Inc. Library of Congress Cataloging In Publication Data: (Revised for volume 3) Lednicer, Daniel, 1929- The organic chemistry of drug synthesis. "A Wiley-lnterscience publication." Includes bibliographical references and index. 1. Chemistry, Pharmaceutical. 2. Drugs. 3. Chemistry, Organic—Synthesis. I. Mitscher, Lester A., joint author. II. Title. [DNLM 1. Chemistry, Organic. 2. Chemistry, Pharmaceutical. 3. Drugs—Chemical synthesis. QV 744 L473o 1977] RS403.L38 615M9 76-28387 ISBN 0-471-09250-9 (v. 3) Printed in the United States of America 10 907654321 With great pleasure we dedicate this book, too, to our wives, Beryle and Betty. The great tragedy of Science is the slaying of a beautiful hypothesis by an ugly fact. Thomas H. Huxley, "Biogenesis and Abiogenisis" Preface Ihe first volume in this series represented the launching of a trial balloon on the part of the authors. In the first place, wo were not entirely convinced that contemporary medicinal (hemistry could in fact be organized coherently on the basis of organic chemistry. -

(12) Patent Application Publication (10) Pub. No.: US 2006/00094.65 A1 Edgar Et Al
US 2006OOO9465A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2006/00094.65 A1 Edgar et al. (43) Pub. Date: Jan. 12, 2006 (54) TREATMENT OF SLEEP DSORDERS USING Related U.S. Application Data SLEEPTARGET MODULATORS (60) Provisional application No. 60/349,912, filed on Jan. 18, 2002. Provisional application No. 60/357,320, (76) Inventors: Dale Edgar, Wayland, MA (US); David filed on Feb. 15, 2002. G. Hangauer, East Amherst, NY (US); Harry Jefferson Leighton, Rockport, Publication Classification ME (US) (51) Int. Cl. A61K 31/496 (2006.01) Correspondence Address: C07D 471/04 (2006.01) MINTZ, LEVIN, COHN, FERRIS, GLOWSKY (52) U.S. Cl. ...................................... 514/253.04; 544/362 AND POPEO, PC. ONE FINANCIAL CENTER (57) ABSTRACT BOSTON, MA 02111 (US) The invention is directed to compositions used for treating Sleep disorders. In addition, the invention provides conve Appl. No.: 10/501,855 nient methods of treatment of a sleep disorder. Furthermore, (21) the invention provides methods of treating Sleep disorders using compositions that remain active for a discrete period (22) PCT Fed: Jan. 21, 2003 of time to reduce side effects. More specifically, the inven tion is directed to the compositions and use of ester deriva tized traZOdone compounds for the treatment of Sleep dis (86) PCT No.: PCT/US03/01845 orders. Patent Application Publication Jan. 12, 2006 Sheet 1 of 8 US 2006/00094.65 A1 IGHRIQ?INH Patent Application Publication Jan. 12, 2006 Sheet 2 of 8 US 2006/00094.65 A1 ZCHRI[10][H Patent Application Publication Jan. 12, 2006 Sheet 3 of 8 US 2006/00094.65 A1 $GIRI[10IH cHEETSWERIN N nOH ad W&N (%) Patent Application Publication Jan. -

Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DIX to the HTSUS—Continued
20558 Federal Register / Vol. 60, No. 80 / Wednesday, April 26, 1995 / Notices DEPARMENT OF THE TREASURY Services, U.S. Customs Service, 1301 TABLE 1.ÐPHARMACEUTICAL APPEN- Constitution Avenue NW, Washington, DIX TO THE HTSUSÐContinued Customs Service D.C. 20229 at (202) 927±1060. CAS No. Pharmaceutical [T.D. 95±33] Dated: April 14, 1995. 52±78±8 ..................... NORETHANDROLONE. A. W. Tennant, 52±86±8 ..................... HALOPERIDOL. Pharmaceutical Tables 1 and 3 of the Director, Office of Laboratories and Scientific 52±88±0 ..................... ATROPINE METHONITRATE. HTSUS 52±90±4 ..................... CYSTEINE. Services. 53±03±2 ..................... PREDNISONE. 53±06±5 ..................... CORTISONE. AGENCY: Customs Service, Department TABLE 1.ÐPHARMACEUTICAL 53±10±1 ..................... HYDROXYDIONE SODIUM SUCCI- of the Treasury. NATE. APPENDIX TO THE HTSUS 53±16±7 ..................... ESTRONE. ACTION: Listing of the products found in 53±18±9 ..................... BIETASERPINE. Table 1 and Table 3 of the CAS No. Pharmaceutical 53±19±0 ..................... MITOTANE. 53±31±6 ..................... MEDIBAZINE. Pharmaceutical Appendix to the N/A ............................. ACTAGARDIN. 53±33±8 ..................... PARAMETHASONE. Harmonized Tariff Schedule of the N/A ............................. ARDACIN. 53±34±9 ..................... FLUPREDNISOLONE. N/A ............................. BICIROMAB. 53±39±4 ..................... OXANDROLONE. United States of America in Chemical N/A ............................. CELUCLORAL. 53±43±0 -
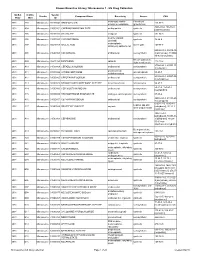
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM -

(12) Patent Application Publication (10) Pub. No.: US 2004/0152713 A1 Petrie (43) Pub
US 2004O152713A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0152713 A1 Petrie (43) Pub. Date: Aug. 5, 2004 (54) METHODS FORTREATING JOINT PAIN OR (52) U.S. Cl. ...................... 514/266.2, 514/310; 514/314; IMPROVING SLEEP USING AN ESTROGEN 514/256 AGONISTIANTAGONIST (57) ABSTRACT (75) Inventor: Charles D. Petrie, Cranston, RI (US) The present invention provides methods, pharmaceutical Correspondence Address: compositions and kits for treating joint pain and/or improv PFIZER INC. ing sleep using a SERM of formula (I): PATENT DEPARTMENT, MS8260-1611 EASTERN POINT ROAD GROTON, CT 06340 (US) (I) (73) Assignee: Pfizer Inc. (21) Appl. No.: 10/761,672 (22) Filed: Jan. 21, 2004 Related U.S. Application Data (60) Provisional application No. 60/441,830, filed on Jan. 22, 2003. Publication Classification wherein the variables A, B, D, E, e, G, Y and Z are as (51) Int. Cl.' .................... A61K 31/517; A61K 31/4709 defined in the Specification. US 2004/0152713 A1 Aug. 5, 2004 METHODS FOR TREATING JOINT PAIN OR ceSS is associated with changes to an individual's circadian IMPROVING SLEEP USING AN ESTROGEN and diurnal rhythms. With increasing age the total amount of AGONISTIANTAGONIST Sleep tends to shorten and the amount of deep sleep can decrease or disappear. Sleep may become more fragmented FIELD OF THE INVENTION and interrupted for the elderly and these changes with timing 0001. This invention relates to methods for treating joint and Structure of Sleep are often associated with Significant pain and/or improving sleep using an estrogen agonist/ morbidity. Similarly, non-elderly individuals may also antagonist also known as Selective estrogen receptor modu exhibit disturbances in the normal Sleep process. -

Imaging Mitochondrial Dynamics in the Adult Heart
Imaging Mitochondrial Dynamics in the Adult Heart Thesis submitted by Siavash Beikoghli Kalkhoran BSc (First class Hons.), MSc (Distinction) For the degree of Doctor of Philosophy University College London, UK. Institute of Cardiovascular Science The Hatter Cardiovascular Institute, University College London, 67 Chenies Mews, London, WC1E 6HX. October 2017 Declaration I, Siavash Beikoghli Kalkhoran, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. The assistance and contribution of individuals to the generation of results are acknowledged within the methods sections of each chapter. 2 Dedicated to Soheila & Taher 3 Abstract Background Mitochondrial dynamics, the phenomenon which incorporates inter-mitochondrial communication and changes in mitochondrial morphology is central to cellular homeostasis. Although the phenomenon of mitochondrial dynamics has been comprehensively studied under normal and pathological conditions in non-cardiac cells, and more recently in cardiac cell lines, its relevance to adult cardiomyocytes has not been so well-established and is investigated in this thesis. Methods and Results Using 2D and 3D electron microscopy, we initially evaluated the morphological features of the 3 different mitochondrial subpopulations (interfibrillar, peri-nuclear, subsarcolemmal) in adult rodent cardiomyocytes, and demonstrated that they are morphologically unique. These morphological characteristics were found to be altered under pathological conditions such as ischaemia or the genetic ablation of mitochondrial fusion proteins “mitofusins”. Using mice expressing the Dendra2 fluorescence probe, we then confirmed that mitochondrial fusion events (“the inter- mitochondrial communication”) occur in live adult cardiomyocytes, and the fusion rates differ according to the mitochondrial subpopulation. -

Mitigating the Inhibition of Human Bile Salt Export Pump by Drugs
DMD Fast Forward. Published on September 7, 2012 as DOI: 10.1124/dmd.112.047068 DMD FastThis Forward. article has not Published been copyedited on andSeptember formatted. The 7, final 2012 version as doi:10.1124/dmd.112.047068may differ from this version. DMD #47968 Mitigating the inhibition of human Bile Salt Export Pump by drugs: opportunities provided by physicochemical property modulation, in-silico modeling and structural modification Daniel J. Warner, Hongming Chen, Louis-David Cantin, J. Gerry Kenna, Simone Stahl, Clare L. Walker, Tobias Noeske. Department of Medicinal Chemistry, AstraZeneca R&D Montreal, Montreal, Quebec, H4S Downloaded from 1Z9, Canada (DJW, LDC) Computational Sciences, Discovery Sciences, AstraZeneca R&D Mölndal, Pepparedsleden dmd.aspetjournals.org 1, Mölndal 43183, Sweden (HC) Molecular Toxicology, Global Safety Assessment, AstraZeneca, Alderley Park, Macclesfield, Cheshire, SK10 4TG, UK (JGK, SS, CLW) Global Safety Assessment, AstraZeneca R&D Mölndal, Pepparedsleden 1, Mölndal 43183, at ASPET Journals on October 10, 2021 Sweden (TN) 1 Copyright 2012 by the American Society for Pharmacology and Experimental Therapeutics. DMD Fast Forward. Published on September 7, 2012 as DOI: 10.1124/dmd.112.047068 This article has not been copyedited and formatted. The final version may differ from this version. DMD #47968 Inhibition of the human Bile Salt Export Pump by drugs. Corresponding author: Tobias Noeske Global Safety Assessment AstraZeneca R&D Mölndal S-431 83 Mölndal, Sweden Phone: +46-31-7064002 Mobile: +46-727-158344