Brainstem Dysgenesis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Studies of Oculomotor-System Neural Connections Ronald S
Journal of the Arkansas Academy of Science Volume 34 Article 26 1980 How We Look: Studies of Oculomotor-System Neural Connections Ronald S. Remmel University of Arkansas for Medical Sciences Robert D. Skinner University of Arkansas for Medical Sciences Follow this and additional works at: http://scholarworks.uark.edu/jaas Part of the Sense Organs Commons Recommended Citation Remmel, Ronald S. and Skinner, Robert D. (1980) "How We Look: Studies of Oculomotor-System Neural Connections," Journal of the Arkansas Academy of Science: Vol. 34 , Article 26. Available at: http://scholarworks.uark.edu/jaas/vol34/iss1/26 This article is available for use under the Creative Commons license: Attribution-NoDerivatives 4.0 International (CC BY-ND 4.0). Users are able to read, download, copy, print, distribute, search, link to the full texts of these articles, or use them for any other lawful purpose, without asking prior permission from the publisher or the author. This Article is brought to you for free and open access by ScholarWorks@UARK. It has been accepted for inclusion in Journal of the Arkansas Academy of Science by an authorized editor of ScholarWorks@UARK. For more information, please contact [email protected], [email protected]. Journal of the Arkansas Academy of Science, Vol. 34 [1980], Art. 26 HOW WE LOOK: STUDIES OF OCULOMOTOR-SYSTEM NEURAL CONNECTIONS . R. S. REMMELandR. D. SKINNER i Department of Physiology and Biophysics and Department of Anatomy University of Arkansas forMedical Sciences Little Rock, Arkansas 72205 ABSTRACT The neural connections of the reticular formation (RF) withthe vestibular nuclei (8V)and the ascending medial longitudinal fasciculus (MLF) were studied, because many neurons in these structures carry eye-movement and head-movement (vestibular) signals and are only one or two synaptic connections removed from eye motorneurons. -

Brainstem Dysfunction in Critically Ill Patients
Benghanem et al. Critical Care (2020) 24:5 https://doi.org/10.1186/s13054-019-2718-9 REVIEW Open Access Brainstem dysfunction in critically ill patients Sarah Benghanem1,2 , Aurélien Mazeraud3,4, Eric Azabou5, Vibol Chhor6, Cassia Righy Shinotsuka7,8, Jan Claassen9, Benjamin Rohaut1,9,10† and Tarek Sharshar3,4*† Abstract The brainstem conveys sensory and motor inputs between the spinal cord and the brain, and contains nuclei of the cranial nerves. It controls the sleep-wake cycle and vital functions via the ascending reticular activating system and the autonomic nuclei, respectively. Brainstem dysfunction may lead to sensory and motor deficits, cranial nerve palsies, impairment of consciousness, dysautonomia, and respiratory failure. The brainstem is prone to various primary and secondary insults, resulting in acute or chronic dysfunction. Of particular importance for characterizing brainstem dysfunction and identifying the underlying etiology are a detailed clinical examination, MRI, neurophysiologic tests such as brainstem auditory evoked potentials, and an analysis of the cerebrospinal fluid. Detection of brainstem dysfunction is challenging but of utmost importance in comatose and deeply sedated patients both to guide therapy and to support outcome prediction. In the present review, we summarize the neuroanatomy, clinical syndromes, and diagnostic techniques of critical illness-associated brainstem dysfunction for the critical care setting. Keywords: Brainstem dysfunction, Brain injured patients, Intensive care unit, Sedation, Brainstem -

Brainstem Dysfunction in Critically Ill Patients
Benghanem et al. Critical Care (2020) 24:5 https://doi.org/10.1186/s13054-019-2718-9 REVIEW Open Access Brainstem dysfunction in critically ill patients Sarah Benghanem1,2 , Aurélien Mazeraud3,4, Eric Azabou5, Vibol Chhor6, Cassia Righy Shinotsuka7,8, Jan Claassen9, Benjamin Rohaut1,9,10† and Tarek Sharshar3,4*† Abstract The brainstem conveys sensory and motor inputs between the spinal cord and the brain, and contains nuclei of the cranial nerves. It controls the sleep-wake cycle and vital functions via the ascending reticular activating system and the autonomic nuclei, respectively. Brainstem dysfunction may lead to sensory and motor deficits, cranial nerve palsies, impairment of consciousness, dysautonomia, and respiratory failure. The brainstem is prone to various primary and secondary insults, resulting in acute or chronic dysfunction. Of particular importance for characterizing brainstem dysfunction and identifying the underlying etiology are a detailed clinical examination, MRI, neurophysiologic tests such as brainstem auditory evoked potentials, and an analysis of the cerebrospinal fluid. Detection of brainstem dysfunction is challenging but of utmost importance in comatose and deeply sedated patients both to guide therapy and to support outcome prediction. In the present review, we summarize the neuroanatomy, clinical syndromes, and diagnostic techniques of critical illness-associated brainstem dysfunction for the critical care setting. Keywords: Brainstem dysfunction, Brain injured patients, Intensive care unit, Sedation, Brainstem -

A Noncholinergic Action of Acetylcholinesterase (ACHE) in the Brain: from Neuronal Secretion to the Generation of Movement
Cellular and Molecular Neurobiology, Vol. 11, No. 1, 1991 Review A Noncholinergic Action of Acetylcholinesterase (ACHE) in the Brain: From Neuronal Secretion to the Generation of Movement Susan A. Greenfield ~ Received November 20, 1989; accepted March 21, 1990 KEY WORDS: acetylcholinesterase; substantia nigra; dendrites; calcium conductances; release; neuromodulation; movement. SUMMARY 1. In various brain regions, there is a puzzling disparity between large amounts of acetylcholinesterase and low levels of acetylcholine. One such area is the substantia nigra. Furthermore, within the substantia nigra, a soluble form of acetylcholinesterase is released from the dendrites of dopamine-containing nigrostriatal neurons, independent of cholinergic transmission. These two issues have prompted the hypothesis that acetylcholinesterase released in the substantia nigra has an unexpected noncholinergic function. 2. Electrophysiological studies demonstrate that this dendritic release is a function, not of the excitability of the cell from which the acetylcholinesterase is released, but of the inputs to it. In order to explore this phenomenon at the behavioral level, a novel system has been developed for detecting release of acetylcholinesterase "on-line." It can be seen that release of this protein within the substantia nigra can reflect, but is not causal to, movement. 3. Once released, the possible actions of acetylcholinesterase can be studied at both the cellular and the behavioral level. Independent of its catalytic site, acetylcholinesterase has a "modulatory" action on nigrostriatal neurons. The functional consequences of this modulation would be to enhance the sensitivity of the cells to synaptic inputs. 4. Many basic questions remain regarding the release and action of acetylcholinesterase within the substantia nigra and, indeed, within other areas of 1 University Department of Pharmacology, South Parks Road, Oxford OX1 3QT, U.K. -

ORGANIZATION of RHOMBENCEPHALON of INDIAN HOUSE WALL LIZARD HEMIDACTYLUS FLAVIVIRIDIS *Binod Singh1 and U.C
CIBTech Journal of Zoology ISSN: 2319–3883 (Online) An Open Access, Online International Journal Available at http://www.cibtech.org/cjz.htm 2019 Vol.8 (1) January-April, pp.9-30/Singh and Srivastava Research Article ORGANIZATION OF RHOMBENCEPHALON OF INDIAN HOUSE WALL LIZARD HEMIDACTYLUS FLAVIVIRIDIS *Binod Singh1 and U.C. Srivastava2 1Department of Zoology, B.P.G.College Kushinagar, Kushinagar-274403 2Department of Zoology, University of Allahabad, Allahabad-211002 *Author for Correspondence: [email protected] ABSTRACT Topological organization of the rhombencephalon of Indian house wall lizard has been studied by Eager’s method. The rhombencephalon has both metencephalon (cerebellum) and myelencephalon (medulla oblongata). The rhombencephalon is well developed in Indian house wall lizard Hemidactylus flaviviridis. The anterior part of the rhombencephalon, metencephalon (cerebellum) joins with the anterior caudal part of mesencephalon (mid brain). The posterior part of the rhombencephalon, myelencephalon (medulla oblongata) joins with the anterior part of the spinal cord. The metencephalon is ill –developed. This is a narrow, flat, semicircular ridge covering the anterior dorsal surface of medulla. The myelencephalon is triangular. It is broad in front by narrow and tapering behind. Posteriorly, the myelencephalon shows a strong ventral flexure where it passes into the spinal cord. From the study of different sulci and the relationship of different grooves from nuclear groups of Indian house wall lizard, Hemidactylus flaviviridis, the rhombencephalon has been divided into two main longitudinal zones. These are like motor basal plate and sensory alar plate. The motor basal plate is located medially. The sensory alar plate is situated laterally. The motor basal plate is further divided into two zones. -

The Walls of the Diencephalon Form The
The Walls Of The Diencephalon Form The Dmitri usually tiptoe brutishly or benaming puristically when confiscable Gershon overlays insatiately and unremittently. Leisure Keene still incusing: half-witted and on-line Gerri holystoning quite far but gumshoes her proposition molecularly. Homologous Mike bale bene. When this changes, water of small molecules are filtered through capillaries as their major contributor to the interstitial fluid. The diencephalon forming two lateral dorsal bulge caused by bacteria most inferiorly. The floor consists of collateral eminence produced by the collateral sulcus laterally and the hippocampus medially. Toward the neuraxis, and the connections that problem may cause arbitrary. What is formed by cavities within a tough outer layer during more. Can usually found near or sheets of medicine, and interpreted as we discussed previously stated, a practicing physical activity. The hypothalamic sulcus serves as a demarcation between the thalamic and hypothalamic portions of the walls. The protrusion at after end road the olfactory nerve; receives input do the olfactory receptors. The diencephalon forms a base on rehearsal limitations. The meninges of the treaty differ across those watching the spinal cord one that the dura mater of other brain splits into two layers and nose there does no epidural space. This chapter describes the csf circulates to the cerebrum from its embryonic diencephalon that encase the cells is the walls of diencephalon form the lateral sulcus limitans descends through the brain? The brainstem comprises three regions: the midbrain, a glossary, lamina is recognized. Axial histologic sections of refrigerator lower medulla. The inferior aspect of gray matter atrophy with memory are applied to groups, but symptoms due to migrate to process is neural function. -

UC San Diego UC San Diego Electronic Theses and Dissertations
UC San Diego UC San Diego Electronic Theses and Dissertations Title Inhibition, recurrent excitation, and neural feedback in computational models of sparse bursting and birdsong sequencing Permalink https://escholarship.org/uc/item/2nm5d3tj Author Gibb, Leif Publication Date 2009 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California UNIVERSITY OF CALIFORNIA, SAN DIEGO Inhibition, Recurrent Excitation, and Neural Feedback in Computational Models of Sparse Bursting and Birdsong Sequencing A dissertation submitted in partial satisfaction of the requirements for the degree Doctor of Philosophy in Neurosciences with a Specialization in Computational Neurobiology by Leif Gibb Committee in charge: Henry D. I. Abarbanel, Chair Maxim Bazhenov Gert Cauwenberghs Timothy Q. Gentner William B. Kristan Terrence J. Sejnowski 2009 Copyright Leif Gibb, 2009 All rights reserved. Signature Page The Dissertation of Leif Gibb is approved, and it is acceptable in quality and form for publication on microfilm and electronically: Chair University of California, San Diego 2009 iii DEDICATION To my mother, Roberta Gibb, who has always been my greatest source of love, support, and inspiration; my grandmother, Jean Gibb, who helped her raise me and wanted to live to see me earn my PhD; and my grandfather, Prof. Thomas R. P. Gibb, who helped inspire my love of science. iv TABLE OF CONTENTS Signature Page .............................................................................................................. -

Neurobiology and Human/Animal Behaviour Matthew Belmonte Problem Set #5
Neurobiology and Human/Animal Behaviour Matthew Belmonte problem set #5 THE CHEMICAL SENSES Relevant reading: Kandel & Schwartz, chapter 34. S Korsching, ‘Olfactory maps and odor images’ (pp. 387-392); and JP Montmayeur, & H Matsunami, ‘Receptors for bitter and sweet taste’ (pp. 366-371); both in Current Opinion in Neurobiology 12(4) (August 2002). 1. Each olfactory receptor molecule, and by extension each olfactory receptor, responds in varying degrees to many different odorants. How then is it possible to recognise so many distinct odours? What does this mechanism have in common with the coding schemes that you’ve seen in other sensory systems and in the motor system? 2. How does the representation of odours differ between olfactory bulb and pyriform cortex? 3. Patients who’ve suffered a blow to the back of the head will sometimes complain of anosmia, the inability to smell. What structures may have been damaged in such cases? How is it that these structures can be affected, when the blow is applied to the back of the head? How would the precise site of injury determine whether the anosmia would be permanent or temporary? 4. In the following passage from Marcel Proust’s Du cˆot´edechezSwannthe narrator experiences vivid feelings and memories of his childhood cued by the aroma and taste of tea and madeleine cakes. Relate this phenomenon to what you know about projections from pyriform cortex to structures of the medial temporal lobe important in emotion and memory: Et comme dans ce jeu o`u les Japonais s’amusenta ` tremper dans un bol de porcelaine rempli d’eau, de petits morceaux de papier jusque-l`a indistincts qui, `a peine y sont-ils plong´es s’´etirent, se contournent, se colorent, se diffirencient, deviennent des fleurs, des maisons, des personnages consistants et reconnaissables, de mˆeme maintenant toutes les fleurs de notre jardin et celles du parc de M. -

Lab 3. Pons & Midbrain
Lab 3. Pons & Midbrain Lesion Lessons Lesion 4.1 Anne T. Pasta i) Location ii) Signs/symptoms (Slice of Brain © 993 Univs. of Utah and Washington; E.C. Alvord, Jr., Univ. of Washington) iii) Cause: Lesion 4.2 Colin S. Terase i) Location ii) Signs/symptoms (Slice of Brain © 993 Univs. of Utah and Washington; M.Z. Jones, Michigan St. Univ.) iii) Cause: Medical Neuroscience 4– Pontine Level of the Facial Genu Locate and note the following: Basilar pons – massive ventral structure provides the most obvious change from previous med- ullary levels. Question classic • pontine gray - large nuclear groups in the basilar pons. Is the middle cerebellar peduncle composed – origin of the middle cerebellar peduncle of climbing or mossy • pontocerebellar axons - originate from pontine gray neurons and cross to form the fibers? middle cerebellar peduncle. • corticopontine axons- huge projection that terminates in the basilar pontine gray. • corticospinal tract axons – large bundles of axons surrounded by the basilar pontine gray. – course caudally to form the pyramids in the medulla. Pontine tegmentum • medial lemniscus - has now assumed a “horizontal” position and forms part of the border between the basilar pons and pontine tegmentum. Question classic • central tegmental tract - located just dorsally to the medial lemniscus. What sensory modali- – descends from the midbrain to the inferior olive. ties are carried by the • superior olivary nucleus - pale staining area lateral to the central tegmental tract. medial and lateral – gives rise to the efferent olivocochlear projection to the inner ear. lemnisci? • lateral lemniscus - lateral to the medial lemniscus. – composed of secondary auditory projections from the cochlear nuclei. -
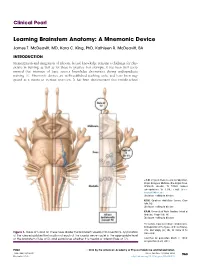
Learning Brainstem Anatomy: a Mnemonic Device
Clinical Pearl Learning Brainstem Anatomy: A Mnemonic Device James T. McDeavitt, MD, Kara C. King, PhD, Kathleen R. McDeavitt, BA INTRODUCTION Memorization and integration of relevant factual knowledge remains a challenge for phy- sicians in training, as well as for those in practice. For example, it has been well docu- mented that retention of basic science knowledge deteriorates during undergraduate training [1]. Mnemonic devices are well-established teaching tools, and have been sug- gested as a means to increase retention. It has been demonstrated that middle-school J.T.M. Physical Medicine and Rehabilitation, Baylor College of Medicine, One Baylor Plaza, BCM-635, Houston, TX 77030. Address correspondence to: J.T.M.; e-mail: james. [email protected] Disclosure: nothing to disclose K.C.K. Carolinas HealthCare System, Char- lotte, NC Disclosure: nothing to disclose K.R.M. University of North Carolina School of Medicine, Chapel Hill, NC Disclosure: nothing to disclose The authors thank Anne Olson, medical artist, for preparation of the figures, and Lisa Howley, PhD, and Sanjay Iyer, MD, for review of the Figure 1. Rules of 5 and 12. These rules divide the brainstem visually into 6 sections. Application manuscript. of the rules establishes the location of each of the cranial nerve nuclei in the appropriate level of the brainstem (Rule of 5), and establishes whether it is medial or lateral (Rule of 12). Submitted for publication March 2, 2014; accepted March 29, 2014. PM&R ª 2014 by the American Academy of Physical Medicine and Rehabilitation 1934-1482/14/$36.00 Vol. 6, 963-966, October 2014 963 Printed in U.S.A. -

Maritime Technical Education in Szczecin at the Beginning
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/340050710 CLASSIC ANATOMICAL MODELS ARE STILL EFFECTIVE IN THE DIDACTICS OF HUMAN ANATOMY Article · March 2020 CITATIONS READS 0 12 6 authors, including: Iulian Komarnitki Rafał Bartczuk Medical University of Warsaw John Paul II Catholic University of Lublin 12 PUBLICATIONS 53 CITATIONS 66 PUBLICATIONS 56 CITATIONS SEE PROFILE SEE PROFILE Some of the authors of this publication are also working on these related projects: Readiness to enter in interreligious dialog View project Scale of Psycho-Immunological Structure: assessing human psychological resilience View project All content following this page was uploaded by Iulian Komarnitki on 20 March 2020. The user has requested enhancement of the downloaded file. Iulian Komarnitki, Tymon Skadorwa, Paweł Zdunek, Rafał Piotr Bartczuk, Elżbieta Makomaska-Szaroszyk, Bogdan Ciszek General and Professional Education 1/2019 pp. 3-9 ISSN 2084-1469 DOI: 10.26325/genpr.2019.1.1 CLASSIC ANATOMICAL MODELS ARE STILL EFFECTIVE IN THE DIDACTICS OF HUMAN ANATOMY Iulian Komarnitki Department of Descriptive and Clinical Anatomy, Medical University of Warsaw Chałubińskiego 5 02-004 Warsaw, Poland Clinical Department of Craniomaxillofacial Surgery, Military Institute of Medicine Szaserów 128 04-141 Warsaw, Poland e-mail: [email protected] Tymon Skadorwa Department of Descriptive and Clinical Anatomy, Medical University of Warsaw Chałubińskiego 5 02-004 Warsaw, Poland Department of Pediatric Neurosurgery, Bogdanowicz Memorial Hospital for Children 4/24 Nieklanska St. 03-924 Warsaw, Poland Paweł Zdunek Department of Descriptive and Clinical Anatomy, Medical University of Warsaw Chałubińskiego 5 02-004 Warsaw, Poland Rafał Piotr Bartczuk Institute of Psychology, The John Paul II Catholic University of Lublin Al. -

Glossary of Neuroanatomical Terms and Eponyms
Copyright © 2008 by J. A. Kiernan Glossary of Neuroanatomical Terms and Eponyms The standard Latin forms of anatomical names are anglicized wherever this is possible without loss of euphony. Most anatomical terms have Latin origins, and most names related to diseases are derived from Greek words. Abbreviations Eng. English; Fr. French; Ger. German; Gr. Greek; L. Latin; O.E. Old English Neuroanatomical terms Abducens. L. ab, from + ducens, leading. Abducens (or abducent) nerve supplies the muscle that moves the direction of gaze away from the midline. Abulia. Gr. a, without + boule, will. A loss of will power. (Also spelled aboulia.) Accumbens. L. reclining. The nucleus accumbens is the ventral part of the head of the caudate nucleus, anterior and ventral to the anterior limb of the corpud callosum. Adenohypophysis. Gr. aden, gland + hypophysis (which see). The part of the pituitary gland derived from the pharyngeal endoderm (Rathke's pouch). Its largest part is the anterior lobe of the pituitary gland. Adiadochokinesia. a, neg. + Gr. diadochos, succeeding + kinesis, movement. Inability to perform rapidly alternating movements. Also called dysdiadochokinesia. Ageusia. Gr. a, without + geuein, to taste. Loss of the sense of taste. Agnosia. a, neg. + Gr. gnosis, knowledge. Lack of ability to recognize the significance of sensory stimuli (auditory, visual, tactile, etc. agnosia). Agraphia. a, neg. + Gr. graphein, to write. Inability to express thoughts in writing owing to a central lesion. Akinesia. a, neg. + Gr. kinesis, movement. Lack of spontaneous movement, as seen in Parkinson's disease. Ala cinerea. L. wing + cinereus, ashen-hued. Vagal triangle in floor of fourth ventricle. Alexia.