Method 1631, Revision E: Mercury in Water by Oxidation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectrometry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effects on Bromine, Chlorine and SO2 Under Air Firing and Oxy-Fuel
GAS-PHASE MERCURY OXIDATION: EFFECTS OF BROMINE, CHLORINE AND SO2 UNDER AIR FIRING AND OXY-FUEL CONDITIONS, EXPERIMENTAL AND MODELING STUDY by Paula Andrea Buitrago A dissertation submitted to the faculty of The University of Utah in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Chemical Engineering The University of Utah August 2011 Copyright © Paula Andrea Buitrago 2011 All Rights Reserved STATEMENT OF DISSERTATION APPROVAL The dissertation of Paula Andrea Bnitrago has been approved by the following supervisory committee members: Geoffrey D. Silcox , Chair 4/1112011 Date Approved JoAnn S. Lighty , Member 4/1112011 Date Approved Jost Wendt , Member 4/18/2011 bate Approved Connie Senior , Member 4/15/2011 Date Approved Eric G. Eddings , Member 4/1112011 bate Approved and by JoAnn S. Lighty , Chair of ----------------~~~~==~~--------------- the Department of Chemical Engineering and by Charles A. Wight, Dean of The Graduate School. ABSTRACT The mercury in coal is emitted in its elemental state when the coal is burned. As the combustion flue gas cools, reactions under homogeneous and heterogeneous conditions between mercury and species such chlorine, bromine, SOx and NOx can take place. The temperature and concentration of these species determines the extent of mercury oxidation. The main objective of this study was to evaluate the effects of bromine, chlorine, SOx and NOx on gas-phase mercury oxidation reactions in flue gas. This study used a methane-fired, bench-scale reactor and CHEMKIN software for performing kinetic calculations. The model included reaction pathways to account for halogen, mercury, and combustion chemistry. The experimental results showed that chlorine is not effective as a gas-phase oxidant of mercury compared with other halogens such as bromine. -

EFGS-SOP-137-R02 and CV-AFS (EPA Method 1631, Rev E)
Document Title: Eurofins Document Reference: Mercury in Water by Oxidation, Purge & Trap EFGS-SOP-137-R02 and CV-AFS (EPA Method 1631, Rev E) Eurofins Document Reference EFGS-SOP-137-R02 Revision 2 Effective Date 6/17/2013 Status Final Historical/Local Document Number FGS-SOP-137.02 Local Document Level Level 3 Local Document Type SOP Local Document Category NA Prepared by Ryan Nelson Reviewed Dave Wunderlich and Patrick Garcia-Strickland and Approved by Revision: 2 Effective Date: 6/17/2013 Page 1 of 20 COMPANY CONFIDENTIAL. All Eurofins Frontier Global Sciences standard operating procedures contain proprietary information and are protected by Washington State Law. Proprietary information must be maintained with the strictest of confidence and must not be used or appropriated to benefit any party without prior written consent from Eurofins Frontier Global Sciences. Document Title: Eurofins Document Reference: Mercury in Water by Oxidation, Purge & Trap EFGS-SOP-137-R02 and CV-AFS (EPA Method 1631, Rev E) Table of Contents 1 Revision Log: ......................................................................................................................... 4 2 Reference: ............................................................................................................................. 4 3 Cross Reference: ................................................................................................................... 5 4 Purpose: ............................................................................................................................... -
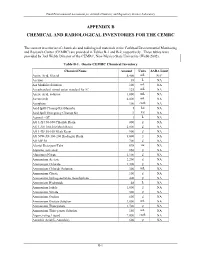
Appendix B Chemical and Radiological Inventories for the Cemrc
Final Environmental Assessment for Actinide Chemistry and Repository Science Laboratory APPENDIX B CHEMICAL AND RADIOLOGICAL INVENTORIES FOR THE CEMRC The current inventories of chemicals and radiological materials at the Carlsbad Environmental Monitoring and Research Center (CEMRC) are provided in Tables B-1 and B-2, respectively. These tables were provided by Joel Webb, Director of the CEMRC, New Mexico State University (Webb 2002). Table B-1. Onsite CEMRC Chemical Inventory Chemical Name Amount Units SARA Limit Acetic Acid, Glacial 5,400 mL NAa Acetone 38 L NA AA Modifier Solution 100 mL NA AccuStandard mixed anion standard for IC 125 mL NA Acetic Acid, solution 1,000 mL NA Acetonitrile 4,000 mL NA Acetylene 100 cu.ft. NA Acid Spill Cleanup Kit (Hazorb) 1 kit NA Acid Spill Emergency Cleanup Kit 3 kit NA Aerosol - OT 1 L NA AG 1-X4 50-100 Chloride Resin 800 g NA AG 1-X8 100-200 Mesh Resin 2,000 g NA AG 1-X8 50-100 Mesh Resin 900 g NA AG 50W-X8 100-200 Hydrogen Resin 3,000 g NA AG MP 50 700 g NA Alcojet Detergent/Tabs 698 oz NA Alumina, activated 850 g NA Aluminum Nitrate 2,100 g NA Ammonium Acetate 2,200 g NA Ammonium Chloride 1,300 g NA Ammonium Chloride Solution 300 mL NA Ammonium Citrate 100 g NA Ammonium hydrogenoxalate, hemihydrate 400 g NA Ammonium Hydroxide 48 L NA Ammonium Iodide 1,000 g NA Ammonium Nitrate 500 g NA Ammonium Oxalate 600 g NA Ammonium Oxalate Solution 2,000 mL NA Ammonium Thiocyanate 1,700 g NA Ammonium Thiocyanate Solution 150 mL NA Argon, refrig. -
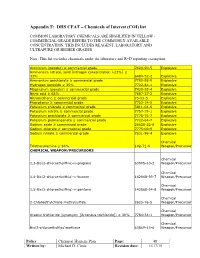
Appendix F: DHS CFAT – Chemicals of Interest (COI) List
Appendix F: DHS CFAT – Chemicals of Interest (COI) list COMMON LABORATORY CHEMICALS ARE HIGHLITED IN YELLOW - COMMERCIAL GRADE REFERS TO THE COMMONLY AVAILABLE CONCENTRATION, THIS INCLUDES REAGENT, LABORATORY AND ULTRAPURE OR HIGHER GRADES Note: This list excludes chemicals under the laboratory and R+D reporting exemption. Aluminum (powder) ≥ commercial grade 7429-90-5 Explosive Ammonium nitrate, solid [nitrogen concentration >23%] ≥ 33% 6484-52-2 Explosive Ammonium perchlorate ≥ commercial grade 7790-98-9 Explosive Hydrogen peroxide ≥ 35% 7722-84-1 Explosive Magnesium (powder) ≥ commercial grade 7439-95-4 Explosive Nitric acid ≥ 68% 7697-37-2 Explosive Nitromethane ≥ commercial grade 75-52-5 Explosive Phosphorus ≥ commercial grade 7723-14-0 Explosive Potassium chlorate ≥ commercial grade 3811-04-9 Explosive Potassium nitrate ≥ commercial grade 7757-79-1 Explosive Potassium perchlorate ≥ commercial grade 7778-74-7 Explosive Potassium permanganate ≥ commercial grade 7722-64-7 Explosive Sodium azide ≥ commercial grade 26628-22-8 Explosive Sodium chlorate ≥ commercial grade 7775-09-9 Explosive Sodium nitrate ≥ commercial grade 7631-99-4 Explosive Chemical Triethanolamine ≥ 80% 102-71-6 Weapon/Precursor CHEMICAL WEAPON/PRECURSORS Chemical 1,3-Bis(2-chloroethylthio)-n-propane 63905-10-2 Weapon/Precursor Chemical 1,4-Bis(2-chloroethylthio)-n-butane 142868-93-7 Weapon/Precursor Chemical 1,5-Bis(2-chloroethylthio)-n-pentane 142868-94-8 Weapon/Precursor Chemical 2-Chloroethylchloro-methylsulfide 2625-76-5 Weapon/Precursor Chemical Arsenic trichloride -

Language of Chemistry 6
CHAPTER Language of Chemistry 6 Section 6.1 Classification of Compounds 2. Compound Classification Compound Classification (a) NH3 binary molecular (b) Fe2O3 binary ionic (c) BaSO4 ternary ionic (d) H2SO4(aq) ternary acid 4. Ion Classification Ion Classification + 2+ (a) NH4 polyatomic cation (b) Sr monoatomic cation 2– 2– (c) S monoatomic anion (d) SO4 polyatomic anion Section 6.2 Monoatomic Ions 6. Monoatomic Cation Stock System Name (a) Ba2+ barium ion (b) Zn2+ zinc ion (c) Co3+ cobalt(III) ion (d) Cu+ copper(I) ion 8. Monoatomic Cation Chemical Formula (a) lithium ion Li+ (b) silver ion Ag+ (c) iron(III) ion Fe3+ (d) tin(IV) ion Sn4+ 10. Monoatomic Cation Latin System Name (a) Cu+ cuprous ion (b) Fe3+ ferric ion (c) Sn4+ stannic ion (d) Pb4+ plumbic ion 2014 © Pearson Education, Inc. Language of Chemistry 33 12. Monoatomic Cation Chemical Formula 2+ (a) mercurous ion Hg2 (b) mercuric ion Hg2+ 14. Monoatomic Anion Systematic Name (a) O2– oxide ion (b) S2– sulfide ion (c) N3– nitride ion (d) P3– phosphide ion Section 6.3 Polyatomic Ions 16. Oxyanion Systematic Name 2– (a) CO3 carbonate ion – (b) HCO3 hydrogen carbonate ion 2– (c) SO4 sulfate ion – (d) HSO4 hydrogen sulfate ion 18. Polyatomic Ion Chemical Formula – (a) chlorate ion ClO3 – (b) perchlorate ion ClO4 – (c) permanganate ion MnO4 – (d) acetate ion C2H3O2 Section 6.4 Writing Chemical Formulas 20. Constituent Ions Chemical Formula (a) Li+ + Cl– LiCl + 2– (b) 2 Ag + O Ag2O 3+ – (c) Cr + 3 I CrI3 2+ 3– (d) 3 Sn + 2 N Sn3N2 22. -

Nitrogen Tribromide Polar Or Nonpolar
Nitrogen tribromide polar or nonpolar Continue Nitrogen Tribromid Names IUPAC Name Nitrogen Tribromid Identifiers CAS Number 15162-90-0 3D Model (JSmol) Interactive Image ChemSpider 20480821 PubChem CID 3082084 CompTox Dashboard (EPA) DTXSID901648 In22 InChI InChIBrH.N/h3'1H;/p-3 SMILES N(Br)(Br)Br Properties Chemical Formula NBr3 Molar Mass 253.7187 g/mol Appearance Deep-red solid melting point explodes at -100 degrees Celsius, except when otherwise noted, the data is given for materials in their standard state (at 25 degrees Celsius), 100 kPa). Infobox links nitrogen tribromid is a chemical compound with the NBr3 formula. It is extremely explosive in its purest form, even at 100 degrees Celsius, and was not isolated until 1975. It's deep-red and volatile solid. The drug NBr3 was first prepared by the reaction of bistrimetligrilbramamin (bis (trimethylsil)amin bromide) with bromine monochloride (with trimethylylyl chloride as a by-product) at 87 degrees on the following equations: (Me3Si)2NBr2 BrCl → NBr3 and 2 Me3SiCl, Where Me is He instantly reacts with ammonia in a dichloromethane solution at 87 degrees Celsius to give NBrH2. Links - Lide, David R. (1998), Handbook on Chemistry and Physics (87 Ed.), Boca-Raton, Florida: CRC Press, p. 4-73, ISBN 0-8493-0594-2 Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of elements (2nd st. Butterworth-Keinmann. page 439. ISBN 978-0-08-037941-8. vteSalts and covalent derivatives of nitrid ion NH3N2H4 He (N2)11 Li3N Be3N2 BN β-C3N4g- C3N4CxNy N2 NxOy NF3 Ne Na3N Mg3 NN2 AlN Si3N4 PNP3N5 SxNySNS4N4 NCl3 Ar K3N Ca3N2 ScN VN CrNCr2N MnxNy FexNy Ni3N CuN n3N2 GaN Ge3N4 as Se NBr3 Kr Rb3 Yn Sr3N2 yn srn NbN β-Mo2N Tc Ru Rh PdN AgN CdN InN Sb Te NI3 Xe Cs3N Ba3N2 Hf3N4 TaN WN Re Os Au Hg3N2 TlN Pb BiN Po At At Rn Fr3N Ra3N2 Rf Db Sg Bh Hs Mt D rg Cn Nh Fl Mc Lv Ts Og s La CeN Pr Nd Pm Sm Eu GdN Tb Dy Er Tm Yb Lu Ac Th Pa UN Np Pu Am Cm Bk Cf Es Fm No Lr Lr Extracted from the Is NBr3 (Nitrogen Tribromid) polar or non-polar? NBr3 (Nitrogen Tribromid) is a polar I'll tell you the polar or nonpolar list below. -

12) United States Patent 10) Patent No.: US 7,173,129 B2
US007 173129B2 12) United States Patent 10) Patent No.: US 7,173,1299 9 B2 Worsencroft et al. (45) Date of Patent: Feb. 6, 2007 (54) SULFONAMIDE-SUBSTITUTED CHALCONE 5,811,449 A 9, 1998 Medford et al. DERVATIVES AND THEIR USE TO TREAT 5,821,260 A 10/1998 Medford et al. DISEASES 5,846,959 A 12/1998 Medford et al. 5,877,203 A 3, 1999 Medford et al. (75) Inventors: Kimberly J. Worsencroft, Alpharetta, 5,951,841 A 9/1999 Wehlage et al. GA (US); Liming Ni, Duluth, GA (US); 6,046,212 A 4/2000 Zwaagstra et al. Zhihong Ye, Lilburn, GA (US); 6,423,740 B1 7/2002 Bombardelli et al. Charleso, Meng, Alpharetta, GA 6,462,075 B1 10/2002 Bowen et al. (US); M. David Weingarten s 6,608, 101 B1 8/2003 Ni et al. s 2003/0232877 A1 12/2003 Sikorski et al. SWISERus) 2003/0236298 Al 12/2003 Meng et al. SE A. Sikorski Atlanta GA (US) 2004/0048858 A1 3/2004 Sikorski et al. (73) Assignee: Athero Genics, Inc., Alpharetta, GA FOREIGN PATENT DOCUMENTS US (US) EP 307762 3, 1989 (*) Notice: Subject to any disclaimer, the term of this EP O 271 307 1, 1992 patent is extended or adjusted under 35 JP 63.01.0720 T 1986 U.S.C. 154(b) by 360 days. JP O4217621 10, 1990 JP O6092950 9, 1992 (21) Appl. No.: 10/863,115 JP 06116206 10, 1992 JP O733O814 6, 1994 (22) Filed: Jun. 7, 2004 WO WO95/1576O 6, 1995 (65) Prior Publication Data (Continued) US 2005/0049236A1 Mar. -
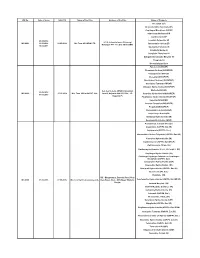
WC No. Date of Issue Valid Till Name of the Firm Address of The
WC No. Date of Issue Valid Till Name of the Firm Address of the Firm Name of Products Irbesartan (EP) Desfesoterodine Succinate (IH) Clopidogrel Bisulphate USP/EP Olmesartan Medoxomil IH Clarithromycin EP 24.05.2016 Losartan Potassium EP Q-1-4, Industrial area ,Ghinrongi WC-0001 13.06.2016 14.05.2019 M/s Teva API INDIA LTD Atorvastatin Calcium EP Malanpur -477 117, Dist -Bhind (MP) 16.02.2017 Quetiapine Fumarate IH Sitagliptin Malate IH Sitagliptin Phosphate IH Dabigatrante Etexilate Mesylate IH Ticagrelor IH Desvenlafaxine Base Famciclovir( IH/USP) Fluvastatin Sodium (IH/USP/EP) Olanzapine(IH/ USP/EP) Irbesartan( IH/USP/EP) Montelukast Sodium(( IH/USP/EP) Quetiapine Fumarate( IH/USP) Diltiazem Hydrochloride(IH/USP/EP) A-2, A-2/1, A-2/2, UPSIDC Industrial Ezetimibe(IH/USP) 22.08.2016 WC-0002 27.05.2019 M/s. Teva API India PVT. Ltd., Area-II, Gajraula-244 235, Dist., J.P. Sertraline Hydrochloride(IH/USP/EP) 07.03.2018 Nagar (U.P) Pioglitazone Hydrochloride(IH/USP/EP) Valsartan(IH/USP/EP) Losartan Potassium(IH/USP/EP) Pregabalin(IH/USP/EP) Atorvastatin Calcium(IH/USP) Caspofungin Acetate(IH) Eletriptan Hydrobromide (IH) Rosuvastatin Calcium (IH/EP) Acamprosate Calcium (Ph. Eur.) Anastrozole (USP/Ph. Eur./IH) Aripiprazole (USP/Ph. Eur.) Atorvastatin Calcium Trihydrate (USP/Ph. Eur./JP) Cinacalcet Hydrochloride (IH) Clarithromycin (USP/Ph. Eur./BP/JP) Clarithromycin Citrate (IH) Clarithromycin Granules 27.5%, 33% & 42% (IH) Clopidogrel Hydrochloride (IH) Clopidogrel Hydrogen Sulphate or Clopidogrel Bisulphate (USP/Ph. Eur.) Colesevelam Hydrochloride (USP) Dapoxetine Hydrochloride (IH) Donepezil Hydrochloride (USP/Ph. Eur./IH) Dutasteride (Ph. -

Alternative Drinking-Water Disinfectants
Alternative drinking-water disinfectants Bromine, iodine and silver Alternative drinking-water disinfectants: Bromine, iodine, silver Alternative drinking-water disinfectants: bromine, iodine and silver ISBN 978-92-4-151369-2 © World Health Organization 2018 Some rights reserved. This work is available under the Creative Commons Attribution- NonCommercial-ShareAlike 3.0 IGO licence (CC BY-NC-SA 3.0 IGO; https://creativecommons.org/licenses/by-nc-sa/3.0/igo). Under the terms of this licence, you may copy, redistribute and adapt the work for non-commercial purposes, provided the work is appropriately cited, as indicated below. In any use of this work, there should be no suggestion that WHO endorses any specific organization, products or services. The use of the WHO logo is not permitted. If you adapt the work, then you must license your work under the same or equivalent Creative Commons licence. If you create a translation of this work, you should add the following disclaimer along with the suggested citation: “This translation was not created by the World Health Organization (WHO). WHO is not responsible for the content or accuracy of this translation. The original English edition shall be the binding and authentic edition”. Any mediation relating to disputes arising under the licence shall be conducted in accordance with the mediation rules of the World Intellectual Property Organization. Suggested citation. Alternative drinking-water disinfectants: bromine, iodine and silver. Geneva: World Health Organization; 2018. Licence: CC BY-NC-SA 3.0 IGO. Cataloguing-in-Publication (CIP) data. CIP data are available at http://apps.who.int/iris. Sales, rights and licensing. -

A Table of Rotational Constants of Symmetric Top Molecules Giving Rise
JOURNAL OF RESEARCH of the National Bureau of Standards - A. Physics and Chemistry Val. 73A, No.2, Marc h- April 1969 A Table of Rotational Constants of Symmetric Top Molecules Givi n9 Rise to Microwave Spectra* Matthew S. Lojko Environmental Science Services Administration, Boulder, Colorado 80302 and Yardley Beers National Bureau of Standards, Boulder Colorado 80302 (December 23, 1968) This paper li sts, in order of increasin g value, the " B" rot ati onal constants of most of the linear and symmetri c top molecules whic h have been observed by mi crowave s pectroscopy. Also are listed the microwave spectral lines which have been observed for the asymmetric tops water and formalde hyde. These d ata are useful for making a quick selection of a molecul e whi ch has a s pectral line close to some previously selected freque ncy. Key words: Asymmetric, linear, and symmetric top molecules; "B" rotationa l constant s; formal dehyde; mi c rowave spectra; s pectral lines; wa ter. I ( Nowadays the re are a number of actual and poten give n in eq (l) it is possible to get a very good approxi tial si tuations in whic h one would lik e to select a mation of th e frequencies of nearly all of the rotational mol ecul e to have a spectral line close to some pre lin es of these molecules. The freque ncy l viously specifi e d frequency_ For one thing, the trans i tions of li ght linear or symme tric top molecul es, whic h j = 2Bj, (1 ) I li e in almost but not exact harmonic relationship, are where } is any integer, whi ch is th e quantum number used to tune up millimeter wave spectrographs_ For gi ving the total angular mome ntum (not in cluding t another, there is a great interest in de vi sing ex peri nuclear spin ) of th e uppe r state giving rise to the transi ments, many involving lasers, in which a spectral line, tion. -

WO 2015/150957 Al 8 October 2015 (08.10.2015) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/150957 Al 8 October 2015 (08.10.2015) P O P C T (51) International Patent Classification: (74) Agent: OLSON, A. Dean; Pfizer Worldwide Research & C07D 471/04 (2006.01) A61P 25/28 (2006.01) Development, Eastern Point Road MS8260-2141, Groton, A61K 31/4985 (2006.01) Connecticut 06340 (US). (21) International Application Number: (81) Designated States (unless otherwise indicated, for every PCT/IB20 15/05 1988 kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, (22) International Filing Date: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, 18 March 2015 (18.03.2015) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (25) Filing Language: English HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (26) Publication Language: English MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (30) Priority Data: PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, 61/973,436 1 April 2014 (01.04.2014) US SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (71) Applicant: PFIZER INC. [US/US]; 235 East 42nd Street, New York, New York 10017 (US). -

United States Patent Office Patented Jan
3,366,622 United States Patent Office Patented Jan. 30, 1968 2 3,366,622 phenyl) and aralkyl having from 1-5 carbon atoms in OX DATION OF HYDRAZOAM DES TO the alkyl chain (for example naphthyl ethyl, but prefer AZOFORMAMDES ably benzyl) groups. John Minton Chalinor and Neil William Woodrow Acording to a preferred embodiment of the invention Porter, Loughborough, England, assignors to Whif 5 R, R, R8 and R4 are each hydrogen. fen & Sons Limited, Loughborough, England The bromine or bromide may be present in amounts No Drawing. Filed July 27, 1965, Ser. No. 475,244 varying over a wide range, but essentially only requires Claims priority, application Great Britain, Aug. 11, 1964, to be present in catalytic amounts. The amount of bro 32,761/64; Oct. 22, 1964, 43,045/64 mine or bromide suitably comprises from 0.1 to 20% 10 Claims. (Cl. 260-192) O by Weight based on the hydrazoamide starting material, and preferably comprises 1 to 10% by weight on the hydrazoamide starting material. Bromides which may be ABSTRACT OF THE DISCLOSURE mentioned include alkali metal bromides, such as for A process for the oxidation of an hydrazoamide, such example sodium bromide and potassium bromide, alkaline as hydrazoformamide to azoformamide, in high yield 5 earth metal bromides, such as for example calcium bro without contamination by the starting material, which mide and barium bromide; other metal bromides such process comprises oxidizing the hydrazoamide with a as for example ferric bromide, chromic bromide, copper water-soluble chlorate in an aqueous acidic medium hav bromide, strontium bromide, cobaltic bromide and zinc ing a pH in the range of 2.0 to 0.5 in the presence of bromide; hydrobromic acid; and halogenobromides such a catalytic amount of bromide or bromine.