Assessment Report
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enzymatic Assay of L-METHIONINE GAMMA-LYASE (EC 4.4.1.11)
Enzymatic Assay of L-METHIONINE GAMMA-LYASE (EC 4.4.1.11) PRINCIPLE: L-Methionine Gamma-Lyase L-Methionine > Methanethiol + 2-Ketobutyrate + NH3 2-Ketobutyrate + MBTH > Azine Derivative Abbreviation used: MBTH = 3-Methyl-2-Benzothiazolinone CONDITIONS: T = 37°C, pH = 8.0, A320nm, Light path = 1 cm METHOD: Stopped Spectrophotometric Rate Determination REAGENTS: A. 100 mM Potassium Phosphate Buffer with 25 mM L-Methionine and 0.01 mM Pyridoxal 5-Phosphate, pH 8.0 at 37°C1 (Prepare 100 ml in deionized water using Potassium Phosphate, Monobasic, Anhydrous, Sigma Prod. No. P-5379, L-Methionine, Sigma Prod. No. M-9625, and Pyridoxal 5-Phosphate, Sigma Prod. No. P-9255. Adjust to pH 8.0 at 37°C with 5 M KOH.) B. 50% (w/v) Trichloroacetic Acid Solution (TCA) (Prepare 5 ml in deionized water using Trichloroacetic Acid, 6.1 N Solution, approximately 100% (w/v), Sigma Stock No. 490-10.) C. 1 M Sodium Acetate Buffer, pH 5.0 at 37°C (NaOAC) (Prepare 100 ml in deionized water using Sodium Acetate, Trihydrate, Sigma Prod. No. S-8625. Adjust to pH 5.0 at 37°C with 5 M HCl.) D. 0.1% (w/v) 3-Methyl-2-Benzothiazolinone Hydrazone (MBTH) (Prepare 10 ml in deionized water using 3-Methyl-2-Benzothiazolinone Hydrazone, Hydrochloride Hydrate, Sigma Prod. No. M-8006.) SSMETH01 Page 1 of 4 07/29/98 Enzymatic Assay of L-METHIONINE GAMMA-LYASE (EC 4.4.1.11) REAGENTS: E. 100 mM Potassium Phosphate Buffer with 1 mM Ethylenediaminetetraacetic Acid, 0.01% (v/v) 2-Mercaptoethanol and 0.02 mM Pyridoxal 5-Phosphate, pH 7.2 at 37°C (Enzyme Diluent)1 (Prepare 10 ml in deionized water using Potassium Phosphate, Monobasic, Anhydrous, Sigma Prod. -
![Lyxumia Subcutaneous Injection 300 Μg [Non-Proprietary Name] Lixisenatide (JAN*) [Name of Applicant] Sanofi K.K](https://docslib.b-cdn.net/cover/4198/lyxumia-subcutaneous-injection-300-g-non-proprietary-name-lixisenatide-jan-name-of-applicant-sanofi-k-k-324198.webp)
Lyxumia Subcutaneous Injection 300 Μg [Non-Proprietary Name] Lixisenatide (JAN*) [Name of Applicant] Sanofi K.K
Report on the Deliberation Results May 31, 2013 Evaluation and Licensing Division, Pharmaceutical and Food Safety Bureau Ministry of Health, Labour and Welfare [Brand name] Lyxumia Subcutaneous Injection 300 μg [Non-proprietary name] Lixisenatide (JAN*) [Name of applicant] Sanofi K.K. [Date of application] June 11, 2012 [Results of deliberation] In the meeting held on May 24, 2013, the First Committee on New Drugs concluded that the product may be approved and that this result should be reported to the Pharmaceutical Affairs Department of the Pharmaceutical Affairs and Food Sanitation Council. The re-examination period for the product is 8 years, and the drug substance and the drug product are both classified as powerful drugs and the product is not classified as a biological product or a specified biological product. The proposed Japanese brand name should be changed for ensuring medical safety. *Japanese Accepted Name (modified INN) This English version of the Japanese review report is intended to be a reference material to provide convenience for users. In the event of inconsistency between the Japanese original and this English translation, the former shall prevail. The PMDA will not be responsible for any consequence resulting from the use of this English version. Review Report May 7, 2013 Pharmaceuticals and Medical Devices Agency The results of a regulatory review conducted by the Pharmaceuticals and Medical Devices Agency on the following pharmaceutical product submitted for registration are as follows. [Brand name] Lyxumia Subcutaneous -

The Un-Design and Design of Insulin: Structural Evolution
THE UN-DESIGN AND DESIGN OF INSULIN: STRUCTURAL EVOLUTION WITH APPLICATION TO THERAPEUTIC DESIGN By NISCHAY K. REGE Submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy Department of Biochemistry Dissertation Advisor: Dr. Michael A. Weiss CASE WESTERN RESERVE UNIVERSITY August, 2018 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of Nischay K. Rege candidate for the degree of Doctor of Philosophy*. Committee Chair Paul Carey Committee Member Michael Weiss Committee Member Faramarz Ismail-Beigi Committee Member George Dubyak Date of Defense: June 26th, 2018 *We also certify that written approval has been obtained for any proprietary material contained therein. Dedication This thesis is dedicated to my mother, Dipti, whose constant love and faith have never failed, to my father, Kiran, who taught me of the virtue of curiosity, and to my wife, Shipra, whose kindness and companionship have given me enough strength for eight lifetimes. i Table of Contents Dedication ..................................................................................................................................... i Table of Contents ......................................................................................................................... ii List of Tables ............................................................................................................................... v List of Figures ........................................................................................................................... -

Genetic Studies of Leptin Concentrations Implicate Leptin in the Regulation of Early
Page 1 of 93 Diabetes Genetic studies of leptin concentrations implicate leptin in the regulation of early adiposity Hanieh Yaghootkar1,2,3*&, Yiying Zhang4&, Cassandra N Spracklen5, Tugce Karaderi6,7,8,9, Lam Opal Huang10, Jonathan Bradfield11,12, Claudia Schurmann13, Rebecca S Fine14,15,16, Michael H Preuss13, Zoltan Kutalik1,17,18, Laura BL Wittemans6,19, Yingchang Lu13,20, Sophia Metz10, Sara M Willems19, Ruifang Li-Gao21, Niels Grarup10, Shuai Wang22, Sophie Molnos23,24, América A Sandoval-Zárate25, Mike A Nalls26,27, Leslie A Lange28, Jeffrey Haesser29, Xiuqing Guo30, Leo-Pekka Lyytikäinen31,32, Mary F Feitosa33, Colleen M Sitlani34, Cristina Venturini35, Anubha Mahajan6,36, Tim Kacprowski37,38, Carol A Wang22, Daniel I Chasman39,40, Najaf Amin41, Linda Broer42, Neil Robertson6,36, Kristin L Young43, Matthew Allison44, Paul L Auer45, Matthias Blüher46, Judith B Borja47,48, Jette Bork-Jensen10, Germán D Carrasquilla10, Paraskevi Christofidou35, Ayse Demirkan41, Claudia A Doege49, Melissa E Garcia50, Mariaelisa Graff43,51, Kaiying Guo4, Hakon Hakonarson11,52, Jaeyoung Hong22, Yii-Der Ida Chen30, Rebecca Jackson53, Hermina Jakupović10, Pekka Jousilahti54, Anne E Justice55, Mika Kähönen56,57, Jorge R Kizer58,59, Jennifer Kriebel23,24, Charles A LeDuc4, Jin Li60, Lars Lind61, Jian’an Luan19, David Mackey62, Massimo Mangino35,63, Satu Männistö54, Jayne F Martin Carli4, Carolina Medina-Gomez41,42, Dennis O Mook-Kanamori21,64, Andrew P Morris65,6,66, Renée de Mutsert21, Matthias Nauck67,38, Ivana Nedeljkovic41, Craig E Pennell22, Arund D Pradhan39,40, Bruce M Psaty68,69, Olli T Raitakari70,71,72, Robert A Scott19, Tea Skaaby73, Konstantin Strauch74,75, Kent D Taylor30, Alexander Teumer76,38, Andre G Uitterlinden41,42, Ying Wu5, Jie Yao30, Mark Walker77, Kari E North43, Peter Kovacs46, M. -
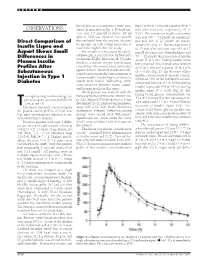
OBSERVATIONS Sured, As Indicated in Fig
LETTERS blood glucose concentrations were mea- lispro at 40 Ϯ 3 min and aspart at 49 Ϯ 3 OBSERVATIONS sured, as indicated in Fig. 1. If blood glu- min after injection, respectively (P ϭ cose was 3.5 mmol/l or lower, 20 ml 0.01). The maximum insulin concentra- glucose 30% was injected. One patient tion was 316 Ϯ 31 pmol/l on insulin lis- Direct Comparison of was excluded from the analysis because, pro and 295 Ϯ 27 pmol/l on insulin by mistake, he took a large extra dose of aspart (NS) (Fig. 1). The increase from 0 Insulin Lispro and insulin the night before the study. to 15 min after injection was 109 Ϯ 17 Aspart Shows Small Free insulin was measured after poly- pmol/l after injection of insulin lispro and Differences in ethylene glycol precipitation by Mercodia 53 Ϯ 11 pmol/l after injection of insulin Iso-Insulin (ELISA; Mercodia AB, Uppsala, Plasma Insulin aspart (P ϭ 0.02). Fasting insulin lispro Sweden), a two-site enzyme immunoassay levels reached 50% of peak concentration Profiles After containing two monoclonal antibodies at 20 Ϯ 1 min and aspart at 30 Ϯ 3 min against insulin. Identical results were ob- (P ϭ 0.02) (Fig. 2). The decrease of free Subcutaneous tained when equimolar concentrations of Injection in Type 1 insulin concentration from peak concen- human insulin, insulin lispro, and insulin tration to 50% of the maximum concen- aspart were tested, indicating 100% Diabetes tration was found at 113 Ϯ 10 min during cross-reactivity between lispro, aspart, insulin lispro and 154 Ϯ 14 min during and human insulin in this assay. -

Thionein Can Serve As a Reducing Agent for the Methionine Sulfoxide Reductases
Thionein can serve as a reducing agent for the methionine sulfoxide reductases Daphna Sagher*†, David Brunell*†, J. Fielding Hejtmancik‡, Marc Kantorow*, Nathan Brot§, and Herbert Weissbach*¶ *Center for Molecular Biology and Biotechnology, Florida Atlantic University, Boca Raton, FL 33431; ‡Ophthalmic Genetic and Visual Function Branch, National Eye Institute, National Institutes of Health, Bethesda, MD 20892; and §Department of Microbiology and Immunology, Hospital for Special Surgery, Cornell University Medical Center, New York, NY 10021 Contributed by Herbert Weissbach, April 7, 2006 It has been generally accepted, primarily from studies on methio- not been reported. Most in vitro studies have used DTT as the nine sulfoxide reductase (Msr) A, that the biological reducing agent reducing agent for both MsrA and MsrB, because this agent for the members of the Msr family is reduced thioredoxin (Trx), works very well with the Msr family of proteins. although high levels of DTT can be used as the reductant in vitro. Recently, the human MsrB genes have been of interest to us as Preliminary experiments using both human recombinant MsrB2 part of our studies on the role of the Msr system in protecting lens (hMsrB2) and MsrB3 (hMsrB3) showed that although DTT can and retinal cells against oxidative damage (18–20). Using a color- function in vitro as the reducing agent, Trx works very poorly, imetric assay for Msr activity, based on the reduction of the prompting a more careful comparison of the ability of DTT and Trx individual epimers of 4-N,N-dimethylaminoazobenzene-4-sulfonyl to function as reducing agents with the various members of the (DABS)-Met(o), we were surprised to find that Trx serves very Msr family. -
![Newborn Screening ACT Sheet [Increased Methionine] Homocystinuria (CBS Deficiency)](https://docslib.b-cdn.net/cover/6249/newborn-screening-act-sheet-increased-methionine-homocystinuria-cbs-deficiency-2586249.webp)
Newborn Screening ACT Sheet [Increased Methionine] Homocystinuria (CBS Deficiency)
American College of Medical Genetics ACT SHEET Newborn Screening ACT Sheet [Increased Methionine] Homocystinuria (CBS Deficiency) Differential Diagnosis: Classical homocystinuria (cystathionine ß-synthase (CBS) deficiency); hypermethioninemia due to methionine adenosyltransferase I/III (MAT I/III) deficiency; glycine n-methyltransferase (GNMT) deficiency; adenosylhomocysteine hydrolase deficiency; liver disease; hyperalimentation. Condition Description: Methionine from ingested protein is normally converted to homocysteine. In classical homocystinuria due to CBS deficiency, homocysteine cannot be converted to cystathionine. As a result, the concentration of homocysteine and its precursor, methionine, will become elevated. In MAT I/III deficiency and the other hypermethioninemias, methionine is increased in the absence of or only with a slightly increased level of homocysteine. YOU SHOULD TAKE THE FOLLOWING ACTIONS: . Contact family to inform them of the newborn screening result and ascertain clinical status. Consult with pediatric metabolic specialist. Evaluate the newborn with attention to liver disease and refer as appropriate. Initiate confirmatory/diagnostic tests in consultation with metabolic specialist. Educate family about homocystinuria and its management, as appropriate. Report findings to newborn screening program. Diagnostic Evaluation: Quantitative plasma amino acids will show increased homocystine and methionine in classical homocystinuria but only increased methionine in the other disorders. Plasma homocysteine analysis will show markedly increased homocysteine in classical homocystinuria and normal or only slightly increased homocysteine in the other disorders. Urine homocysteine is markedly increased in classical homocystinuria. Clinical Considerations: Homocystinuria is usually asymptomatic in the neonate. If untreated, these children eventually develop mental retardation, ectopia lentis, a marfanoid appearance including arachnodactyly, osteoporosis, other skeletal deformities and thromboembolism. MAT I/III deficiency may be benign. -

Transfer of Β-Hydroxy-Β-Methylbutyrate from Sows to Their
Wan et al. Journal of Animal Science and Biotechnology (2017) 8:2 DOI 10.1186/s40104-016-0132-6 RESEARCH Open Access Transfer of β-hydroxy-β-methylbutyrate from sows to their offspring and its impact on muscle fiber type transformation and performance in pigs Haifeng Wan†, Jiatao Zhu†, Caimei Wu†, Pan Zhou, Yong Shen, Yan Lin, Shengyu Xu, Lianqiang Che, Bin Feng, Jian Li, Zhengfeng Fang and De Wu* Abstract Background: Previous studies suggested that supplementation of lactating sows with β-hydroxy-β-methylbutyrate (HMB) could improve the performance of weaning pigs, but there were little information in the muscle fiber type transformation of the offspring and the subsequent performance in pigs from weaning through finishing in response to maternal HMB consumption. The purpose of this study was to determine the effect of supplementing lactating sows with HMB on skeletal muscle fiber type transformation and growth of the offspring during d 28 and 180 after birth. A total of 20 sows according to their body weight were divided into the control (CON, n = 10) or HMB groups (HMB, n = 10). Sows in the HMB group were supplemented with β-hydroxy-β-methylbutyrate calcium (HMB-Ca) 2 g /kg feed during d 1 to 27 of lactation. After weaning, 48 mixed sex piglets were blocked by sow treatment and fed standard diets for post-weaning, growing, finishing periods. Growth performance was recorded during d 28 to 180 after birth. Pigs were slaughtered on d 28 (n = 6/treatment) and 180 (n = 6/treatment) postnatal, and the longissimus dorsi (LD) was collected, respectively. -

MEDICATION GUIDE ADLYXIN (Ad-LIX-In) (Lixisenatide) Injection
MEDICATION GUIDE ADLYXIN (ad-LIX-in) (lixisenatide) injection, for subcutaneous use What is the most important information I should know about ADLYXIN? Do not share your ADLYXIN pen with other people, even if the needle has been changed. You may give other people a serious infection, or get a serious infection from them. ADLYXIN can cause serious side effects including inflammation of the pancreas (pancreatitis), which may be severe and lead to death. Before using ADLYXIN, tell your healthcare provider if you have had: • pancreatitis • a history of alcoholism • stones in your gallbladder (cholelithiasis) These medical problems may make you more likely to get pancreatitis. Stop taking ADLYXIN and call your healthcare provider right away if you have pain in your stomach area (abdomen) that is severe, and will not go away. The pain may be felt going from your abdomen through to your back. The pain may happen with or without vomiting. These may be symptoms of pancreatitis. What is ADLYXIN? ADLYXIN is an injectable prescription medicine that may improve blood sugar (glucose) control in adults with type 2 diabetes, when used with diet and exercise. • ADLYXIN is not insulin. • ADLYXIN is not a substitute for insulin and is not for use in people with type 1 diabetes or people with diabetic ketoacidosis. • ADLYXIN has not been studied in people with a history of pancreatitis. • ADLYXIN has not been studied in people who use short-acting insulin. • ADLYXIN has not been studied in people who have a stomach problem that causes slow emptying of the stomach (gastroparesis). ADLYXIN is not for people with slow emptying of the stomach. -

Networking Breakfast with ASBMR Leaders, NIH Representatives And
8/20/2019 ASBMR Connect: Networking Breakfast with ASBMR Leaders, NIH Representatives and Senior Investigators September 20, 2019, 06:45 AM W308/Orange County Convention Center ASBMR Connect is supported in part by educational grants from Amgen, Inc. and Ultragenyx Pharmaceutical Inc. The Networking Breakfast with ASBMR Leaders, NIH Representatives and Senior Investigators is a ticketed event that is part of the ASBMR Connect Program. ASBMR Connect requires advance registration and a separate ticket fee. Registration Open September 20, 2019, 07:00 AM Valencia Ballroom Lobby/Orange County Convention Center Gerald D. Aurbach Lecture and Presentation of Esteemed Awards September 20, 2019, 08:00 AM Valencia Ballroom B-D/Orange County Convention Center Join your colleagues to celebrate the following ASBMR 2019 Esteemed Award Winners: William F. Neuman Award, Paula Stern Achievement Award, Stephen M. Krane Award and Adele L. Boskey Award From Genes to Genomes to Biology and Health • Richard Lifton MD, PhD, Rockefeller University September 20, 08:30 AM-09:30 AM Valencia Ballroom B-D/Orange County Convention Center Networking Break September 20, 2019, 09:30 AM Valencia Foyer/Orange County Convention Center Highlights of the ASBMR 2019 Annual Meeting September 20, 2019, 10:00 AM Valencia Ballroom B-D/Orange County Convention Center • Dana Gaddy PhD, College of Veterinary Medicine, Texas A&M University, United States, Chair • Shonni Silverberg MD, Columbia University College of Physicians & Surgeons, United States, Chair • Johannes Van Leeuwen PhD, Erasmus University Medical Center, Netherlands, Chair This special session is of interest to all health professionals, first time meeting attendees, young investigators, individuals new to the field, nurses, clinical research study coordinators, physical therapists and/or those seeking guidance in navigating through the extensive ASBMR program. -

Advantages and Disadvantages of Different Treatment Methods in Achondroplasia: a Review
International Journal of Molecular Sciences Review Advantages and Disadvantages of Different Treatment Methods in Achondroplasia: A Review Wiktoria Wrobel, Emilia Pach and Iwona Ben-Skowronek * Metabolic Laboratory, Department of Paediatric Endocrinology and Diabetology with Endocrine, Medical University in Lublin, Prof. A. Gebala Street 6, 20-093 Lublin, Poland; [email protected] (W.W.); [email protected] (E.P.) * Correspondence: [email protected]; Tel.: +48-817-185-440 Abstract: Achondroplasia (ACH) is a disease caused by a missense mutation in the FGFR3 (fibroblast growth factor receptor 3) gene, which is the most common cause of short stature in humans. The treatment of ACH is necessary and urgent because untreated achondroplasia has many complications, both orthopedic and neurological, which ultimately lead to disability. This review presents the current and potential pharmacological treatments for achondroplasia, highlighting the advantages and disadvantages of all the drugs that have been demonstrated in human and animal studies in different stages of clinical trials. The article includes the potential impacts of drugs on achondroplasia symptoms other than short stature, including their effects on spinal canal stenosis, the narrowing of the foramen magnum and the proportionality of body structure. Addressing these effects could significantly improve the quality of life of patients, possibly reducing the frequency and necessity of hospitalization and painful surgical procedures, which are currently the only therapeutic options used. The criteria for a good drug for achondroplasia are best met by recombinant human growth hormone at present and will potentially be met by vosoritide in the future, while the rest of the drugs Citation: Wrobel, W.; Pach, E.; are in the early stages of clinical trials. -

Methionine Metabolism in Chronic Liver Diseases: an Update on Molecular Mechanism and Therapeutic Implication
Signal Transduction and Targeted Therapy www.nature.com/sigtrans REVIEW ARTICLE OPEN Methionine metabolism in chronic liver diseases: an update on molecular mechanism and therapeutic implication Zhanghao Li1, Feixia Wang1, Baoyu Liang1, Ying Su1, Sumin Sun1, Siwei Xia1, Jiangjuan Shao2,3, Zili Zhang1,2,3, Min Hong1, Feng Zhang1,2,3 and Shizhong Zheng1,2,3 As one of the bicyclic metabolic pathways of one-carbon metabolism, methionine metabolism is the pivot linking the folate cycle to the transsulfuration pathway. In addition to being a precursor for glutathione synthesis, and the principal methyl donor for nucleic acid, phospholipid, histone, biogenic amine, and protein methylation, methionine metabolites can participate in polyamine synthesis. Methionine metabolism disorder can aggravate the damage in the pathological state of a disease. In the occurrence and development of chronic liver diseases (CLDs), changes in various components involved in methionine metabolism can affect the pathological state through various mechanisms. A methionine-deficient diet is commonly used for building CLD models. The conversion of key enzymes of methionine metabolism methionine adenosyltransferase (MAT) 1 A and MAT2A/MAT2B is closely related to fibrosis and hepatocellular carcinoma. In vivo and in vitro experiments have shown that by intervening related enzymes or downstream metabolites to interfere with methionine metabolism, the liver injuries could be reduced. Recently, methionine supplementation has gradually attracted the attention of many clinical researchers. Most researchers agree that adequate methionine supplementation can help reduce liver damage. Retrospective analysis of recently conducted relevant studies is of profound significance. This paper reviews the latest achievements related to methionine metabolism and CLD, from molecular 1234567890();,: mechanisms to clinical research, and provides some insights into the future direction of basic and clinical research.