MOCHI Enables Discovery of Heterogeneous Interactome Modules in 3D Nucleome
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ENSG Gene Encodes Effector TCR Pathway Costimulation Inhibitory/Exhaustion Synapse/Adhesion Chemokines/Receptors
ENSG Gene Encodes Effector TCR pathway Costimulation Inhibitory/exhaustion Synapse/adhesion Chemokines/receptors ENSG00000111537 IFNG IFNg x ENSG00000109471 IL2 IL-2 x ENSG00000232810 TNF TNFa x ENSG00000271503 CCL5 CCL5 x x ENSG00000139187 KLRG1 Klrg1 x ENSG00000117560 FASLG Fas ligand x ENSG00000121858 TNFSF10 TRAIL x ENSG00000134545 KLRC1 Klrc1 / NKG2A x ENSG00000213809 KLRK1 Klrk1 / NKG2D x ENSG00000188389 PDCD1 PD-1 x x ENSG00000117281 CD160 CD160 x x ENSG00000134460 IL2RA IL-2 receptor x subunit alpha ENSG00000110324 IL10RA IL-10 receptor x subunit alpha ENSG00000115604 IL18R1 IL-18 receptor 1 x ENSG00000115607 IL18RAP IL-18 receptor x accessory protein ENSG00000081985 IL12RB2 IL-12 receptor x beta 2 ENSG00000186810 CXCR3 CXCR3 x x ENSG00000005844 ITGAL CD11a x ENSG00000160255 ITGB2 CD18; Integrin x x beta-2 ENSG00000156886 ITGAD CD11d x ENSG00000140678 ITGAX; CD11c x x Integrin alpha-X ENSG00000115232 ITGA4 CD49d; Integrin x x alpha-4 ENSG00000169896 ITGAM CD11b; Integrin x x alpha-M ENSG00000138378 STAT4 Stat4 x ENSG00000115415 STAT1 Stat1 x ENSG00000170581 STAT2 Stat2 x ENSG00000126561 STAT5a Stat5a x ENSG00000162434 JAK1 Jak1 x ENSG00000100453 GZMB Granzyme B x ENSG00000145649 GZMA Granzyme A x ENSG00000180644 PRF1 Perforin 1 x ENSG00000115523 GNLY Granulysin x ENSG00000100450 GZMH Granzyme H x ENSG00000113088 GZMK Granzyme K x ENSG00000057657 PRDM1 Blimp-1 x ENSG00000073861 TBX21 T-bet x ENSG00000115738 ID2 ID2 x ENSG00000176083 ZNF683 Hobit x ENSG00000137265 IRF4 Interferon x regulatory factor 4 ENSG00000140968 IRF8 Interferon -

Establishing the Pathogenicity of Novel Mitochondrial DNA Sequence Variations: a Cell and Molecular Biology Approach
Mafalda Rita Avó Bacalhau Establishing the Pathogenicity of Novel Mitochondrial DNA Sequence Variations: a Cell and Molecular Biology Approach Tese de doutoramento do Programa de Doutoramento em Ciências da Saúde, ramo de Ciências Biomédicas, orientada pela Professora Doutora Maria Manuela Monteiro Grazina e co-orientada pelo Professor Doutor Henrique Manuel Paixão dos Santos Girão e pela Professora Doutora Lee-Jun C. Wong e apresentada à Faculdade de Medicina da Universidade de Coimbra Julho 2017 Faculty of Medicine Establishing the pathogenicity of novel mitochondrial DNA sequence variations: a cell and molecular biology approach Mafalda Rita Avó Bacalhau Tese de doutoramento do programa em Ciências da Saúde, ramo de Ciências Biomédicas, realizada sob a orientação científica da Professora Doutora Maria Manuela Monteiro Grazina; e co-orientação do Professor Doutor Henrique Manuel Paixão dos Santos Girão e da Professora Doutora Lee-Jun C. Wong, apresentada à Faculdade de Medicina da Universidade de Coimbra. Julho, 2017 Copyright© Mafalda Bacalhau e Manuela Grazina, 2017 Esta cópia da tese é fornecida na condição de que quem a consulta reconhece que os direitos de autor são pertença do autor da tese e do orientador científico e que nenhuma citação ou informação obtida a partir dela pode ser publicada sem a referência apropriada e autorização. This copy of the thesis has been supplied on the condition that anyone who consults it recognizes that its copyright belongs to its author and scientific supervisor and that no quotation from the -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -
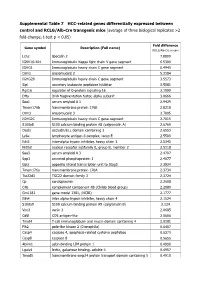
Supplemental Table 7 HCC-Related Genes Differentially Expressed Between Control and RCLG/Alb-Cre Transgenic Mice
Supplemental Table 7 HCC-related genes differentially expressed between control and RCLG/Alb-Cre transgenic mice (average of three biological replicates >2 fold-change, t-test p < 0.05) Fold difference Gene symbol Description (Full name) (RCLG/Alb-Cre vs con) Lcn2 lipocalin 2 7.8899 IGKV16-104 Immunoglobulin Kappa light chain V gene segment 6.5300 IGHG1 Immunoglobulin heavy chain C gene segment 6.4945 Orm2 orosomucoid 2 5.3184 IGHG2B Immunoglobulin heavy chain C gene segment 3.5573 Slpi secretory leukocyte peptidase inhibitor 3.5081 Rgs16 regulator of G-protein signaling 16 3.1999 Dffa DNA fragmentation factor, alpha subunit 3.0666 Saa1 serum amyloid A 1 2.9429 Tmem176b transmembrane protein 176B 2.8218 Orm3 orosomucoid 3 2.7805 IGHG2C Immunoglobulin heavy chain C gene segment 2.7015 S100a8 S100 calcium binding protein A8 (calgranulin A) 2.6769 Ocel1 occludin/ELL domain containing 1 2.6553 Ly6e lymphocyte antigen 6 complex, locus E 2.5509 Itih3 inter-alpha trypsin inhibitor, heavy chain 3 2.5345 Nr0b2 nuclear receptor subfamily 0, group B, member 2 2.5118 Saa3 serum amyloid A 3 2.4707 Spp1 secreted phosphoprotein 1 2.4077 Gats opposite strand transcription unit to Stag3 2.3934 Tmem176a transmembrane protein 176A 2.3734 Tsc22d3 TSC22 domain family 3 2.3724 Cp ceruloplasmin 2.2608 C4b complement component 4B (Childo blood group) 2.2089 Gm1381 gene model 1381, (NCBI) 2.1777 Itih4 inter alpha-trypsin inhibitor, heavy chain 4 2.1524 S100a9 S100 calcium binding protein A9 (calgranulin B) 2.124 Vnn3 vanin 3 2.0685 Cd5l CD5 antigen-like 2.0606 -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid
The Capacity of Long-Term in Vitro Proliferation of Acute Myeloid Leukemia Cells Supported Only by Exogenous Cytokines Is Associated with a Patient Subset with Adverse Outcome Annette K. Brenner, Elise Aasebø, Maria Hernandez-Valladares, Frode Selheim, Frode Berven, Ida-Sofie Grønningsæter, Sushma Bartaula-Brevik and Øystein Bruserud Supplementary Material S2 of S31 Table S1. Detailed information about the 68 AML patients included in the study. # of blasts Viability Proliferation Cytokine Viable cells Change in ID Gender Age Etiology FAB Cytogenetics Mutations CD34 Colonies (109/L) (%) 48 h (cpm) secretion (106) 5 weeks phenotype 1 M 42 de novo 241 M2 normal Flt3 pos 31.0 3848 low 0.24 7 yes 2 M 82 MF 12.4 M2 t(9;22) wt pos 81.6 74,686 low 1.43 969 yes 3 F 49 CML/relapse 149 M2 complex n.d. pos 26.2 3472 low 0.08 n.d. no 4 M 33 de novo 62.0 M2 normal wt pos 67.5 6206 low 0.08 6.5 no 5 M 71 relapse 91.0 M4 normal NPM1 pos 63.5 21,331 low 0.17 n.d. yes 6 M 83 de novo 109 M1 n.d. wt pos 19.1 8764 low 1.65 693 no 7 F 77 MDS 26.4 M1 normal wt pos 89.4 53,799 high 3.43 2746 no 8 M 46 de novo 26.9 M1 normal NPM1 n.d. n.d. 3472 low 1.56 n.d. no 9 M 68 MF 50.8 M4 normal D835 pos 69.4 1640 low 0.08 n.d. -

Serum Albumin OS=Homo Sapiens
Protein Name Cluster of Glial fibrillary acidic protein OS=Homo sapiens GN=GFAP PE=1 SV=1 (P14136) Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 Cluster of Isoform 3 of Plectin OS=Homo sapiens GN=PLEC (Q15149-3) Cluster of Hemoglobin subunit beta OS=Homo sapiens GN=HBB PE=1 SV=2 (P68871) Vimentin OS=Homo sapiens GN=VIM PE=1 SV=4 Cluster of Tubulin beta-3 chain OS=Homo sapiens GN=TUBB3 PE=1 SV=2 (Q13509) Cluster of Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 (P60709) Cluster of Tubulin alpha-1B chain OS=Homo sapiens GN=TUBA1B PE=1 SV=1 (P68363) Cluster of Isoform 2 of Spectrin alpha chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTAN1 (Q13813-2) Hemoglobin subunit alpha OS=Homo sapiens GN=HBA1 PE=1 SV=2 Cluster of Spectrin beta chain, non-erythrocytic 1 OS=Homo sapiens GN=SPTBN1 PE=1 SV=2 (Q01082) Cluster of Pyruvate kinase isozymes M1/M2 OS=Homo sapiens GN=PKM PE=1 SV=4 (P14618) Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 Clathrin heavy chain 1 OS=Homo sapiens GN=CLTC PE=1 SV=5 Filamin-A OS=Homo sapiens GN=FLNA PE=1 SV=4 Cytoplasmic dynein 1 heavy chain 1 OS=Homo sapiens GN=DYNC1H1 PE=1 SV=5 Cluster of ATPase, Na+/K+ transporting, alpha 2 (+) polypeptide OS=Homo sapiens GN=ATP1A2 PE=3 SV=1 (B1AKY9) Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 Dihydropyrimidinase-related protein 2 OS=Homo sapiens GN=DPYSL2 PE=1 SV=1 Cluster of Alpha-actinin-1 OS=Homo sapiens GN=ACTN1 PE=1 SV=2 (P12814) 60 kDa heat shock protein, mitochondrial OS=Homo -

Growth and Molecular Profile of Lung Cancer Cells Expressing Ectopic LKB1: Down-Regulation of the Phosphatidylinositol 3-Phosphate Kinase/PTEN Pathway1
[CANCER RESEARCH 63, 1382–1388, March 15, 2003] Growth and Molecular Profile of Lung Cancer Cells Expressing Ectopic LKB1: Down-Regulation of the Phosphatidylinositol 3-Phosphate Kinase/PTEN Pathway1 Ana I. Jimenez, Paloma Fernandez, Orlando Dominguez, Ana Dopazo, and Montserrat Sanchez-Cespedes2 Molecular Pathology Program [A. I. J., P. F., M. S-C.], Genomics Unit [O. D.], and Microarray Analysis Unit [A. D.], Spanish National Cancer Center, 28029 Madrid, Spain ABSTRACT the cell cycle in G1 (8, 9). However, the intrinsic mechanism by which LKB1 activity is regulated in cells and how it leads to the suppression Germ-line mutations in LKB1 gene cause the Peutz-Jeghers syndrome of cell growth is still unknown. It has been proposed that growth (PJS), a genetic disease with increased risk of malignancies. Recently, suppression by LKB1 is mediated through p21 in a p53-dependent LKB1-inactivating mutations have been identified in one-third of sporadic lung adenocarcinomas, indicating that LKB1 gene inactivation is critical in mechanism (7). In addition, it has been observed that LKB1 binds to tumors other than those of the PJS syndrome. However, the in vivo brahma-related gene 1 protein (BRG1) and this interaction is required substrates of LKB1 and its role in cancer development have not been for BRG1-induced growth arrest (10). Similar to what happens in the completely elucidated. Here we show that overexpression of wild-type PJS, Lkb1 heterozygous knockout mice show gastrointestinal hamar- LKB1 protein in A549 lung adenocarcinomas cells leads to cell-growth tomatous polyposis and frequent hepatocellular carcinomas (11, 12). suppression. To examine changes in gene expression profiles subsequent to Interestingly, the hamartomas, but not the malignant tumors, arising in exogenous wild-type LKB1 in A549 cells, we used cDNA microarrays. -

Triplet Repeat Length Bias and Variation in the Human Transcriptome
Triplet repeat length bias and variation in the human transcriptome Michael Mollaa,1,2, Arthur Delcherb,1, Shamil Sunyaevc, Charles Cantora,d,2, and Simon Kasifa,e aDepartment of Biomedical Engineering and dCenter for Advanced Biotechnology, Boston University, Boston, MA 02215; bCenter for Bioinformatics and Computational Biology, University of Maryland, College Park, MD 20742; cDepartment of Medicine, Division of Genetics, Brigham and Women’s Hospital and Harvard Medical School, Boston, MA 02115; and eCenter for Advanced Genomic Technology, Boston University, Boston, MA 02215 Contributed by Charles Cantor, July 6, 2009 (sent for review May 4, 2009) Length variation in short tandem repeats (STRs) is an important family including Huntington’s disease (10) and hereditary ataxias (11, 12). of DNA polymorphisms with numerous applications in genetics, All Huntington’s patients exhibit an expanded number of copies in medicine, forensics, and evolutionary analysis. Several major diseases the CAG tandem repeat subsequence in the N terminus of the have been associated with length variation of trinucleotide (triplet) huntingtin gene. Moreover, an increase in the repeat length is repeats including Huntington’s disease, hereditary ataxias and spi- anti-correlated to the onset age of the disease (13). Multiple other nobulbar muscular atrophy. Using the reference human genome, we diseases have also been associated with copy number variation of have catalogued all triplet repeats in genic regions. This data revealed tandem repeats (8, 14). Researchers have hypothesized that inap- a bias in noncoding DNA repeat lengths. It also enabled a survey of propriate repeat variation in coding regions could result in toxicity, repeat-length polymorphisms (RLPs) in human genomes and a com- incorrect folding, or aggregation of a protein. -

Functional Mechanisms Underlying Pleiotropic Risk Alleles at the 19P13.1 Breast&Ndash;Ovarian Cancer Susceptibility Locus
ARTICLE Received 16 Jun 2015 | Accepted 20 Jul 2016 | Published 7 Sep 2016 DOI: 10.1038/ncomms12675 OPEN Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast–ovarian cancer susceptibility locus Kate Lawrenson et al.# A locus at 19p13 is associated with breast cancer (BC) and ovarian cancer (OC) risk. Here we analyse 438 SNPs in this region in 46,451 BC and 15,438 OC cases, 15,252 BRCA1 mutation carriers and 73,444 controls and identify 13 candidate causal SNPs associated with serous OC (P ¼ 9.2 Â 10 À 20), ER-negative BC (P ¼ 1.1 Â 10 À 13), BRCA1-associated BC (P ¼ 7.7 Â 10 À 16) and triple negative BC (P-diff ¼ 2 Â 10 À 5). Genotype-gene expression associations are identified for candidate target genes ANKLE1 (P ¼ 2 Â 10 À 3) and ABHD8 (Po2 Â 10 À 3). Chromosome conformation capture identifies interactions between four candidate SNPs and ABHD8, and luciferase assays indicate six risk alleles increased transactivation of the ADHD8 promoter. Targeted deletion of a region containing risk SNP rs56069439 in a putative enhancer induces ANKLE1 downregulation; and mRNA stability assays indicate functional effects for an ANKLE1 30-UTR SNP. Altogether, these data suggest that multiple SNPs at 19p13 regulate ABHD8 and perhaps ANKLE1 expression, and indicate common mechanisms underlying breast and ovarian cancer risk. Correspondence and requests for materials should be addressed to A.C.A. (email: [email protected]). #A full list of authors and their affiliations appears at the end of the paper. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase