Differential Diagnosis of NAFLD- a Short Summary
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of Abnormal Liver Chemistries
ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries Paul Y. Kwo, MD, FACG, FAASLD1, Stanley M. Cohen, MD, FACG, FAASLD2, and Joseph K. Lim, MD, FACG, FAASLD3 1Division of Gastroenterology/Hepatology, Department of Medicine, Stanford University School of Medicine, Palo Alto, California, USA; 2Digestive Health Institute, University Hospitals Cleveland Medical Center and Division of Gastroenterology and Liver Disease, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA; 3Yale Viral Hepatitis Program, Yale University School of Medicine, New Haven, Connecticut, USA. Am J Gastroenterol 2017; 112:18–35; doi:10.1038/ajg.2016.517; published online 20 December 2016 Abstract Clinicians are required to assess abnormal liver chemistries on a daily basis. The most common liver chemistries ordered are serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase and bilirubin. These tests should be termed liver chemistries or liver tests. Hepatocellular injury is defined as disproportionate elevation of AST and ALT levels compared with alkaline phosphatase levels. Cholestatic injury is defined as disproportionate elevation of alkaline phosphatase level as compared with AST and ALT levels. The majority of bilirubin circulates as unconjugated bilirubin and an elevated conjugated bilirubin implies hepatocellular disease or cholestasis. Multiple studies have demonstrated that the presence of an elevated ALT has been associated with increased liver-related mortality. A true healthy normal ALT level ranges from 29 to 33 IU/l for males, 19 to 25 IU/l for females and levels above this should be assessed. The degree of elevation of ALT and or AST in the clinical setting helps guide the evaluation. -

Viral Hepatitis Testing Effective Date: January 1, 2012
Viral Hepatitis Testing Effective Date: January 1, 2012 Scope This guideline provides guidance for the use of laboratory tests to diagnose acute and chronic viral hepatitis in adults (> 19 years) in the primary care setting. General Considerations for Ordering Laboratory Tests Prior to ordering tests for hepatitis, consider the patient’s history, age, risk factors (see below), hepatitis vaccination status, and any available previous hepatitis test results. Risk Factors for Viral Hepatitis include: • Substance use (includes sharing drug snorting, smoking or injection equipment) • High-risk sexual activity or sexual partner with viral hepatitis • Travel to or from high-risk hepatitis endemic areas or exposure during a local outbreak • Immigration from hepatitis B and/or C endemic countries • Household contact with an infected person especially if personal items (e.g., razors, toothbrushes, nail clippers) are shared • Recipient of unscreened blood products* • Needle-stick injury or other occupational exposure (e.g., healthcare workers) • Children born to mothers with chronic hepatitis B or C infection • Attendance at daycare • Contaminated food or water (hepatitis A only) • Tattoos and body piercing • History of incarceration • HIV or other sexually transmitted infection • Hemodialysis *screening of donated blood products for hepatitis C (anti-HCV) began in 1990 in Canada.1 Types of Viral Hepatitis Hepatitis A: causes acute but not chronic hepatitis Hepatitis B: causes acute and chronic hepatitis Hepatitis C: causes chronic hepatitis but rarely manifests as acute hepatitis Hepatitis D: rare and only occurs in patients infected with hepatitis B Hepatitis E: clinically similar to hepatitis A, mostly restricted to endemic areas and occasionally causes chronic infection in immunosuppressed people Others: e.g. -

A Drug-Induced Cholestatic Pattern
Review articles Hepatotoxicity: A Drug-Induced Cholestatic Pattern Laura Morales M.,1 Natalia Vélez L.,1 Octavio Germán Muñoz M., MD.2 1 Medical Student in the Faculty of Medicine and Abstract the Gastrohepatology Group at the Universidad de Antioquia in Medellín, Colombia Although drug induced liver disease is a rare condition, it explains 40% to 50% of all cases of acute liver 2 Internist and Hepatologist at the Hospital Pablo failure. In 20% to 40% of the cases, the pattern is cholestatic and is caused by inhibition of the transporters Tobon Uribe and in the Gastrohepatology Group at that regulate bile synthesis. This reduction in activity is directly or indirectly mediated by drugs and their me- the Universidad de Antioquia in Medellín, Colombia tabolites and/or by genetic polymorphisms and other risk factors of the patient. Its manifestations range from ......................................... biochemical alterations in the absence of symptoms to acute liver failure and chronic liver damage. Received: 30-01-15 Although there is no absolute test or marker for diagnosis of this disease, scales and algorithms have Accepted: 26-01-16 been developed to assess the likelihood of cholestatic drug induced liver disease. Other types of evidence are not routinely used because of their complexity and cost. Diagnosis is primarily based on exclusion using circumstantial evidence. Cholestatic drug induced liver disease has better overall survival rates than other patters, but there are higher risks of developing chronic liver disease. In most cases, the patient’s condition improves when the drug responsible for the damage is removed. Hemodialysis and transplantation should be considered only for selected cases. -

Prevention & Control of Viral Hepatitis Infection
Prevention & Control of Viral Hepatitis Infection: A Strategy for Global Action © World Health Organization 2011. All rights reserved. The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines for which there may not yet be full agreement. The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions excepted, the names of proprietary products are distinguished by initial capital letters. All reasonable precautions have been taken by WHO to verify the information contained in this publication. However, the published material is being distributed without warranty of any kind, either express or implied. The responsibility for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for damages arising from its use. Table of contents Disease burden 02 What is viral hepatitis? 05 Prevention & control: a tailored approach 06 Global Achievements 08 Remaining challenges 10 World Health Assembly: a mandate for comprehensive prevention & control 13 WHO goals and strategy -

Acute Liver Failure J G O’Grady
148 Postgrad Med J: first published as 10.1136/pgmj.2004.026005 on 4 March 2005. Downloaded from REVIEW Acute liver failure J G O’Grady ............................................................................................................................... Postgrad Med J 2005;81:148–154. doi: 10.1136/pgmj.2004.026005 Acute liver failure is a complex multisystemic illness that account for most cases, but a significant number of patients have no definable cause and are evolves quickly after a catastrophic insult to the liver classified as seronegative or of being of indeter- leading to the development of encephalopathy. The minate aetiology. Paracetamol is the commonest underlying aetiology and the pace of progression strongly cause in the UK and USA.2 Idiosyncratic reac- tions comprise another important group. influence the clinical course. The commonest causes are paracetamol, idiosyncratic drug reactions, hepatitis B, and Viral seronegative hepatitis. The optimal care is multidisciplinary ALF is an uncommon complication of viral and up to half of the cases receive liver transplants, with hepatitis, occurring in 0.2%–4% of cases depend- ing on the underlying aetiology.3 The risk is survival rates around 75%–90%. Artificial liver support lowest with hepatitis A, but it increases with the devices remain unproven in efficacy in acute liver failure. age at time of exposure. Hepatitis B can be associated with ALF through a number of ........................................................................... scenarios (table 2). The commonest are de novo infection and spontaneous surges in viral repli- cation, while the incidence of the delta virus cute liver failure (ALF) is a complex infection seems to be decreasing rapidly. multisystemic illness that evolves after a Vaccination should reduce the incidence of Acatastrophic insult to the liver manifesting hepatitis A and B, while antiviral drugs should in the development of a coagulopathy and ameliorate replication of hepatitis B. -

Chronic Viral Hepatitis in a Cohort of Inflammatory Bowel Disease
pathogens Article Chronic Viral Hepatitis in a Cohort of Inflammatory Bowel Disease Patients from Southern Italy: A Case-Control Study Giuseppe Losurdo 1,2 , Andrea Iannone 1, Antonella Contaldo 1, Michele Barone 1 , Enzo Ierardi 1 , Alfredo Di Leo 1,* and Mariabeatrice Principi 1 1 Section of Gastroenterology, Department of Emergency and Organ Transplantation, University “Aldo Moro” of Bari, 70124 Bari, Italy; [email protected] (G.L.); [email protected] (A.I.); [email protected] (A.C.); [email protected] (M.B.); [email protected] (E.I.); [email protected] (M.P.) 2 Ph.D. Course in Organs and Tissues Transplantation and Cellular Therapies, Department of Emergency and Organ Transplantation, University “Aldo Moro” of Bari, 70124 Bari, Italy * Correspondence: [email protected]; Tel.: +39-080-559-2925 Received: 14 September 2020; Accepted: 21 October 2020; Published: 23 October 2020 Abstract: We performed an epidemiologic study to assess the prevalence of chronic viral hepatitis in inflammatory bowel disease (IBD) and to detect their possible relationships. Methods: It was a single centre cohort cross-sectional study, during October 2016 and October 2017. Consecutive IBD adult patients and a control group of non-IBD subjects were recruited. All patients underwent laboratory investigations to detect chronic hepatitis B (HBV) and C (HCV) infection. Parameters of liver function, elastography and IBD features were collected. Univariate analysis was performed by Student’s t or chi-square test. Multivariate analysis was performed by binomial logistic regression and odds ratios (ORs) were calculated. We enrolled 807 IBD patients and 189 controls. Thirty-five (4.3%) had chronic viral hepatitis: 28 HCV (3.4%, versus 5.3% in controls, p = 0.24) and 7 HBV (0.9% versus 0.5% in controls, p = 0.64). -

Zoonotic Diseases Fact Sheet
ZOONOTIC DISEASES FACT SHEET s e ion ecie s n t n p is ms n e e s tio s g s m to a a o u t Rang s p t tme to e th n s n m c a s a ra y a re ho Di P Ge Ho T S Incub F T P Brucella (B. Infected animals Skin or mucous membrane High and protracted (extended) fever. 1-15 weeks Most commonly Antibiotic melitensis, B. (swine, cattle, goats, contact with infected Infection affects bone, heart, reported U.S. combination: abortus, B. suis, B. sheep, dogs) animals, their blood, tissue, gallbladder, kidney, spleen, and laboratory-associated streptomycina, Brucellosis* Bacteria canis ) and other body fluids causes highly disseminated lesions bacterial infection in tetracycline, and and abscess man sulfonamides Salmonella (S. Domestic (dogs, cats, Direct contact as well as Mild gastroenteritiis (diarrhea) to high 6 hours to 3 Fatality rate of 5-10% Antibiotic cholera-suis, S. monkeys, rodents, indirect consumption fever, severe headache, and spleen days combination: enteriditis, S. labor-atory rodents, (eggs, food vehicles using enlargement. May lead to focal chloramphenicol, typhymurium, S. rep-tiles [especially eggs, etc.). Human to infection in any organ or tissue of the neomycin, ampicillin Salmonellosis Bacteria typhi) turtles], chickens and human transmission also body) fish) and herd animals possible (cattle, chickens, pigs) All Shigella species Captive non-human Oral-fecal route Ranges from asymptomatic carrier to Varies by Highly infective. Low Intravenous fluids primates severe bacillary dysentery with high species. 16 number of organisms and electrolytes, fevers, weakness, severe abdominal hours to 7 capable of causing Antibiotics: ampicillin, cramps, prostration, edema of the days. -

Primary Biliary Cholangitis: a Brief Overview Justin S
REVIEW Primary Biliary Cholangitis: A Brief Overview Justin S. Louie,* Sirisha Grandhe,* Karen Matsukuma,† and Christopher L. Bowlus* Primary biliary cholangitis (PBC), previously referred to supported by the higher concordance of PBC in monozy- as primary biliary cirrhosis, is the most common chronic gotic compared with dizygotic twins.4 In addition, certain cholestatic autoimmune disease affecting adults in the human leukocyte antigen haplotypes have been associ- United States.1 It is characterized by a hallmark serologic ated with PBC, as well as variants at loci along the inter- signature, antimitochondrial antibody (AMA), and specific leukin-12 (IL-12) immunoregulatory pathway (IL-12A and bile duct pathology with progressive intrahepatic duct de- IL-12RB2 loci).5 struction leading to cholestasis. PBC is potentially fatal and can have both intrahepatic and extrahepatic complications. PATHOGENESIS EPIDEMIOLOGY The primary disease mechanism in PBC is thought to be T cell lymphocyte–mediated injury against intralobu- PBC affects all races and ethnicities; however, it is best lar biliary epithelial cells. This causes progressive destruc- studied in the Caucasian population. The condition pre- tion and eventual disappearance of the intralobular bile dominantly affects women older than 40 years, with a ducts. Molecular mimicry has been proposed as the ini- female/male ratio of 9:1.2 Although the incidence of PBC tiating event in the loss of tolerance primarily to mito- appears to be stable, the overall prevalence of the disease chondrial pyruvate dehydrogenase complex, E2, during is increasing.3 An individual’s genetic susceptibility, epige- which exogenous antigens evoke an immune response netic factors, and certain environmental triggers seem to that recognizes an endogenous (self) antigen inciting an play important roles. -
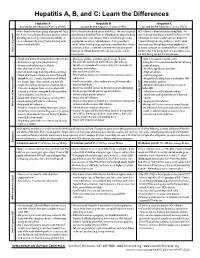
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Active Peptic Ulcer Disease in Patients with Hepatitis C Virus-Related Cirrhosis: the Role of Helicobacter Pylori Infection and Portal Hypertensive Gastropathy
dore.qxd 7/19/2004 11:24 AM Page 521 View metadata, citation and similar papers at core.ac.uk ORIGINAL ARTICLE brought to you by CORE provided by Crossref Active peptic ulcer disease in patients with hepatitis C virus-related cirrhosis: The role of Helicobacter pylori infection and portal hypertensive gastropathy Maria Pina Dore MD PhD, Daniela Mura MD, Stefania Deledda MD, Emmanouil Maragkoudakis MD, Antonella Pironti MD, Giuseppe Realdi MD MP Dore, D Mura, S Deledda, E Maragkoudakis, Ulcère gastroduodénal évolutif chez les A Pironti, G Realdi. Active peptic ulcer disease in patients patients atteints de cirrhose liée au HCV : Le with hepatitis C virus-related cirrhosis: The role of Helicobacter pylori infection and portal hypertensive rôle de l’infection à Helicobacter pylori et de la gastropathy. Can J Gastroenterol 2004;18(8):521-524. gastropathie liée à l’hypertension portale BACKGROUND & AIM: The relationship between Helicobacter HISTORIQUE ET BUT : Le lien entre l’infection à Helicobacter pylori pylori infection and peptic ulcer disease in cirrhosis remains contro- et l’ulcère gastroduodénal dans la cirrhose reste controversé. Le but de la versial. The purpose of the present study was to investigate the role of présente étude est de vérifier le rôle de l’infection à H. pylori et de la gas- H pylori infection and portal hypertension gastropathy in the preva- tropathie liée à l’hypertension portale dans la prévalence de l’ulcère gas- lence of active peptic ulcer among dyspeptic patients with compen- troduodénal évolutif chez les patients dyspeptiques souffrant d’une sated hepatitis C virus (HCV)-related cirrhosis. -

Understanding Human Astrovirus from Pathogenesis to Treatment
University of Tennessee Health Science Center UTHSC Digital Commons Theses and Dissertations (ETD) College of Graduate Health Sciences 6-2020 Understanding Human Astrovirus from Pathogenesis to Treatment Virginia Hargest University of Tennessee Health Science Center Follow this and additional works at: https://dc.uthsc.edu/dissertations Part of the Diseases Commons, Medical Sciences Commons, and the Viruses Commons Recommended Citation Hargest, Virginia (0000-0003-3883-1232), "Understanding Human Astrovirus from Pathogenesis to Treatment" (2020). Theses and Dissertations (ETD). Paper 523. http://dx.doi.org/10.21007/ etd.cghs.2020.0507. This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected]. Understanding Human Astrovirus from Pathogenesis to Treatment Abstract While human astroviruses (HAstV) were discovered nearly 45 years ago, these small positive-sense RNA viruses remain critically understudied. These studies provide fundamental new research on astrovirus pathogenesis and disruption of the gut epithelium by induction of epithelial-mesenchymal transition (EMT) following astrovirus infection. Here we characterize HAstV-induced EMT as an upregulation of SNAI1 and VIM with a down regulation of CDH1 and OCLN, loss of cell-cell junctions most notably at 18 hours post-infection (hpi), and loss of cellular polarity by 24 hpi. While active transforming growth factor- (TGF-) increases during HAstV infection, inhibition of TGF- signaling does not hinder EMT induction. However, HAstV-induced EMT does require active viral replication. -

Hepatitis C 2005 Clinical Guidelines Summary of The: New York State Department of Health Clinical Guidelines for the Medical Management of Hepatitis C
Hepatitis C 2005 Clinical Guidelines Summary of the: New York State Department of Health Clinical Guidelines for the Medical Management of Hepatitis C Inside: Key Features of Viral Hepatitis A,B and C 1 Natural Course of HCV Infection 2 Persons at Risk for HCV Infection 3 Sources of HCV Infection 4 Counseling Prior To Testing 5 Screening for HCV Algorithm 6 Laboratory Testing for HCV 7 Interpretation of HCV Test Results 8 Post Exposure Screening for HCV 9 Counseling After Testing 10 Treating HCV Patients 11 Medical Management of HCV Positive Patients 12 References and Internet Resources 13 Hepatitis C Virus The Hidden Epidemic The Burden of HCV • 3 million Americans are chronically infected with the • 8-10,000 deaths a year are caused by HCV. Hepatitis C virus (HCV). • HCV is the leading cause for liver transplants and • 342,000 New Yorkers are estimated to be infected chronic liver disease. with HCV. • HCV deaths will increase four-fold to 38,000, by • A majority of the people infected with HCV do not the year 2010. know they have it. • Years of life lost to Hepatitis C (2001-2019) • Thousands of people go undetected each year—due 3.1 million years to inadequate risk assessment, under-screening and confusion about the use of diagnostic tests. • Cost of premature disability and death (2010-2109) $75.5 billion Hepatitis C Virus • Direct medical costs in absentee losses due to Hepatitis C $750 million/ year • HCV is a blood-borne disease transmitted by • Total medical expenditures for persons with HCV blood-to-blood contact.