Diagnosis and Management of Primary Biliary Cholangitis Ticle
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

12 Retina Gabriele K
299 12 Retina Gabriele K. Lang and Gerhard K. Lang 12.1 Basic Knowledge The retina is the innermost of three successive layers of the globe. It comprises two parts: ❖ A photoreceptive part (pars optica retinae), comprising the first nine of the 10 layers listed below. ❖ A nonreceptive part (pars caeca retinae) forming the epithelium of the cil- iary body and iris. The pars optica retinae merges with the pars ceca retinae at the ora serrata. Embryology: The retina develops from a diverticulum of the forebrain (proen- cephalon). Optic vesicles develop which then invaginate to form a double- walled bowl, the optic cup. The outer wall becomes the pigment epithelium, and the inner wall later differentiates into the nine layers of the retina. The retina remains linked to the forebrain throughout life through a structure known as the retinohypothalamic tract. Thickness of the retina (Fig. 12.1) Layers of the retina: Moving inward along the path of incident light, the individual layers of the retina are as follows (Fig. 12.2): 1. Inner limiting membrane (glial cell fibers separating the retina from the vitreous body). 2. Layer of optic nerve fibers (axons of the third neuron). 3. Layer of ganglion cells (cell nuclei of the multipolar ganglion cells of the third neuron; “data acquisition system”). 4. Inner plexiform layer (synapses between the axons of the second neuron and dendrites of the third neuron). 5. Inner nuclear layer (cell nuclei of the bipolar nerve cells of the second neuron, horizontal cells, and amacrine cells). 6. Outer plexiform layer (synapses between the axons of the first neuron and dendrites of the second neuron). -

Evaluation of Abnormal Liver Chemistries
ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries Paul Y. Kwo, MD, FACG, FAASLD1, Stanley M. Cohen, MD, FACG, FAASLD2, and Joseph K. Lim, MD, FACG, FAASLD3 1Division of Gastroenterology/Hepatology, Department of Medicine, Stanford University School of Medicine, Palo Alto, California, USA; 2Digestive Health Institute, University Hospitals Cleveland Medical Center and Division of Gastroenterology and Liver Disease, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA; 3Yale Viral Hepatitis Program, Yale University School of Medicine, New Haven, Connecticut, USA. Am J Gastroenterol 2017; 112:18–35; doi:10.1038/ajg.2016.517; published online 20 December 2016 Abstract Clinicians are required to assess abnormal liver chemistries on a daily basis. The most common liver chemistries ordered are serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase and bilirubin. These tests should be termed liver chemistries or liver tests. Hepatocellular injury is defined as disproportionate elevation of AST and ALT levels compared with alkaline phosphatase levels. Cholestatic injury is defined as disproportionate elevation of alkaline phosphatase level as compared with AST and ALT levels. The majority of bilirubin circulates as unconjugated bilirubin and an elevated conjugated bilirubin implies hepatocellular disease or cholestasis. Multiple studies have demonstrated that the presence of an elevated ALT has been associated with increased liver-related mortality. A true healthy normal ALT level ranges from 29 to 33 IU/l for males, 19 to 25 IU/l for females and levels above this should be assessed. The degree of elevation of ALT and or AST in the clinical setting helps guide the evaluation. -

A Drug-Induced Cholestatic Pattern
Review articles Hepatotoxicity: A Drug-Induced Cholestatic Pattern Laura Morales M.,1 Natalia Vélez L.,1 Octavio Germán Muñoz M., MD.2 1 Medical Student in the Faculty of Medicine and Abstract the Gastrohepatology Group at the Universidad de Antioquia in Medellín, Colombia Although drug induced liver disease is a rare condition, it explains 40% to 50% of all cases of acute liver 2 Internist and Hepatologist at the Hospital Pablo failure. In 20% to 40% of the cases, the pattern is cholestatic and is caused by inhibition of the transporters Tobon Uribe and in the Gastrohepatology Group at that regulate bile synthesis. This reduction in activity is directly or indirectly mediated by drugs and their me- the Universidad de Antioquia in Medellín, Colombia tabolites and/or by genetic polymorphisms and other risk factors of the patient. Its manifestations range from ......................................... biochemical alterations in the absence of symptoms to acute liver failure and chronic liver damage. Received: 30-01-15 Although there is no absolute test or marker for diagnosis of this disease, scales and algorithms have Accepted: 26-01-16 been developed to assess the likelihood of cholestatic drug induced liver disease. Other types of evidence are not routinely used because of their complexity and cost. Diagnosis is primarily based on exclusion using circumstantial evidence. Cholestatic drug induced liver disease has better overall survival rates than other patters, but there are higher risks of developing chronic liver disease. In most cases, the patient’s condition improves when the drug responsible for the damage is removed. Hemodialysis and transplantation should be considered only for selected cases. -

Non-Hepatic Hyperammonaemia: an Important, Potentially Reversible Cause of Encephalopathy
Postgrad Med J 2001;77:717–722 717 Postgrad Med J: first published as 10.1136/pmj.77.913.717 on 1 November 2001. Downloaded from CASE REPORTS Non-hepatic hyperammonaemia: an important, potentially reversible cause of encephalopathy N D Hawkes, G A O Thomas, A Jurewicz, O M Williams, C E M Hillier, I N F McQueen, G Shortland Abstract Case reports The clinical syndrome of encephalopathy CASE 1 is most often encountered in the context of A 20 year old man was admitted to a local hos- decompensated liver disease and the diag- pital with two days of inappropriate behaviour, nosis is usually clear cut. Non-hepatic clumsiness, drowsiness, memory loss, slurred causes of encephalopathy are rarer and speech, and abdominal discomfort. Since the tend to present to a wide range of medical age of 2 years he had suVered recurrent rectal specialties with variable and episodic bleeding and investigation had revealed haem- symptoms. Delay can result in the devel- orrhoids. Bleeding from his rectum had contin- opment of potentially life threatening ued over the years but had been worse recently. complications, such as seizures and coma. On examination he was confused. His Early recognition is vital. A history of Glasgow coma scale score (GCS) was 15/15. similar episodes or clinical risk factors Neurological and general examination was nor- and early assessment of blood ammonia mal, with no stigmata of chronic liver disease. levels help establish the diagnosis. In Investigations showed a leucocytosis (leuco- × 9 addition to adequate supportive care, cyte count 22 10 /l) and serum bilirubin level investigation of the underlying cause of of 32 µmol/l. -

Acute Liver Failure J G O’Grady
148 Postgrad Med J: first published as 10.1136/pgmj.2004.026005 on 4 March 2005. Downloaded from REVIEW Acute liver failure J G O’Grady ............................................................................................................................... Postgrad Med J 2005;81:148–154. doi: 10.1136/pgmj.2004.026005 Acute liver failure is a complex multisystemic illness that account for most cases, but a significant number of patients have no definable cause and are evolves quickly after a catastrophic insult to the liver classified as seronegative or of being of indeter- leading to the development of encephalopathy. The minate aetiology. Paracetamol is the commonest underlying aetiology and the pace of progression strongly cause in the UK and USA.2 Idiosyncratic reac- tions comprise another important group. influence the clinical course. The commonest causes are paracetamol, idiosyncratic drug reactions, hepatitis B, and Viral seronegative hepatitis. The optimal care is multidisciplinary ALF is an uncommon complication of viral and up to half of the cases receive liver transplants, with hepatitis, occurring in 0.2%–4% of cases depend- ing on the underlying aetiology.3 The risk is survival rates around 75%–90%. Artificial liver support lowest with hepatitis A, but it increases with the devices remain unproven in efficacy in acute liver failure. age at time of exposure. Hepatitis B can be associated with ALF through a number of ........................................................................... scenarios (table 2). The commonest are de novo infection and spontaneous surges in viral repli- cation, while the incidence of the delta virus cute liver failure (ALF) is a complex infection seems to be decreasing rapidly. multisystemic illness that evolves after a Vaccination should reduce the incidence of Acatastrophic insult to the liver manifesting hepatitis A and B, while antiviral drugs should in the development of a coagulopathy and ameliorate replication of hepatitis B. -

MOC PORT/DOCK Practice Core Modules Content Outline
MOC PORT/DOCK Practice Core Modules Content Outline 1 Cataract and Anterior Segment (10%) 1.1 Basic Anatomical and Scientific Aspects 1.1.1 The Normal Lens 1.1.1.1 Anatomy 1.2 Assessment Techniques 1.2.1 History-Taking and Preoperative Examination of Ocular Structures 1.2.1.1 Take patient history 1.2.1.2 Examine ocular structures 1.2.1.3 Signs and symptoms 1.2.1.4 Indications for surgery 1.2.1.6 External examination 1.2.1.7 Slit-lamp examination 1.2.1.8 Fundus evaluation 1.2.1.9 Impact on visual functioning and quality of life 1.2.2 Measurement of Visual and Macular Function 1.2.2.1 Visual acuity testing 1.2.2.2 Test visual acuity 1.2.2.5 Visual field testing 1.2.2.6 Test visual field 1.2.2.7 Cataract's effect on visual acuity 1.2.2.8 Estimate cataract's effect on visual acuity 1.2.2.10 Measure visual (and macular) function 1.2.4.3 Evaluate glare disability: subjective and objective 1.3 Clinical Disease Conditions and Manifestations 1.3.1 Types of Cataract 1.3.1.1 Identify types of cataracts 1.3.2 Anterior Segment Disorders 1.3.2.1 Diagnose anterior segment disorders 1.3.2.2 Cataract associated with uveitis 1.3.2.3 Diabetes mellitus and cataract formation 1.3.2.4 Lens-induced glaucoma 1.3.2.4.1 Lens particle 1.3.2.4.2 Phacolytic 1.3.2.4.3 Phacomorphic Copyright and Use Policy for ABO Content Outlines © 2017 – American Board of Ophthalmology. -

Picquestion of the Week:3/09/09
McKechnie Field - Spring Training Home of the Pirates PIC QUESTION OF THE WEEK: 3/09/09 Q: Why is rifaximin used in patients with pouchitis? A: Pouchitis is an inflammation of the internal pouch fashioned from small intestinal tissue during ileal pouch anal anastamosis (IPAA). This surgical procedure bypasses the large intestine and may be employed in patients with ulcerative colitis or Crohn’s disease. A temporary ileostomy is placed to allow the pouch to heal without risk of infection. IPAA is considered preferable to an ostomy for patients suffering from inflammatory bowel diseases refractory to medical treatment. The pouch serves as a collection device for waste, but permits the patient to experience regular bowel movements. It is typical for patients with an internal pouch to experience more frequent (average 6 per day) and watery bowel movements. Pouchitis is the most common complication of IPAA and widely regarded as an idiopathic disease; however, colonization by fecal bacteria may be a contributing factor. The condition occurs most commonly in the first six months following reversal of the ileostomy. Although its frequency decreases after six months, nearly 50% of patients with an IPAA will eventually experience pouchitis. Presenting symptoms include diarrhea, increased frequency of bowel movements, bleeding, abdominal pain, and fever. It can result in dehydration and, in severe cases, hospitalization. Treatment of acute pouchitis generally consists of a two-week course of antibiotic therapy with metronidazole or ciprofloxacin. Approximately 10% of patients do not respond to initial treatment and develop chronic (> 4 weeks) pouchitis. A small number of clinical trials support the potential use of rifaximin for the treatment of refractory or recurrent pouchitis. -
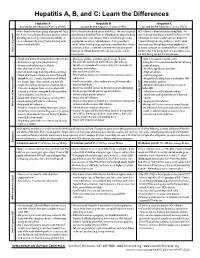
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Skin Manifestations of Liver Diseases
medigraphic Artemisaen línea AnnalsA Koulaouzidis of Hepatology et al. 2007; Skin manifestations6(3): July-September: of liver 181-184diseases 181 Editorial Annals of Hepatology Skin manifestations of liver diseases A. Koulaouzidis;1 S. Bhat;2 J. Moschos3 Introduction velop both xanthelasmas and cutaneous xanthomas (5%) (Figure 7).1 Other disease-associated skin manifestations, Both acute and chronic liver disease can manifest on but not as frequent, include the sicca syndrome and viti- the skin. The appearances can range from the very subtle, ligo.2 Melanosis and xerodermia have been reported. such as early finger clubbing, to the more obvious such PBC may also rarely present with a cutaneous vasculitis as jaundice. Identifying these changes early on can lead (Figures 8 and 9).3-5 to prompt diagnosis and management of the underlying condition. In this pictorial review we will describe the Alcohol related liver disease skin manifestations of specific liver conditions illustrat- ed with appropriate figures. Dupuytren’s contracture was described initially by the French surgeon Guillaume Dupuytren in the 1830s. General skin findings in liver disease Although it has other causes, it is considered a strong clinical pointer of alcohol misuse and its related liver Chronic liver disease of any origin can cause typical damage (Figure 10).6 Therapy options other than sur- skin findings. Jaundice, spider nevi, leuconychia and fin- gery include simvastatin, radiation, N-acetyl-L-cys- ger clubbing are well known features (Figures 1 a, b and teine.7,8 Facial lipodystrophy is commonly seen as alco- 2). Palmar erythema, “paper-money” skin (Figure 3), ro- hol replaces most of the caloric intake in advanced al- sacea and rhinophyma are common but often overlooked coholism (Figure 11). -

Familial Hypercholesterolemia and Xanthomatosis Associated with Diabetes Mellitus: a Case Report and Review of the Literature Ch
Familial Hypercholesterolemia and Xanthomatosis Associated with Diabetes Mellitus: A Case Report and Review of the Literature Ching-Hsiang Leung, Tien-Ling Chen*, Chao-Hung Wang, Kun-Wu Tsan, and Daniel T. H. Chin** Division of Endocrinology and Metabolism, Department of Internal Medicine; *Division of Allergy, Immunology & Rheumatology, Department of Internal Medicine; **Department of Pathology; Mackay Memorial Hospital, Taipei, Taiwan Abstract Familial hypercholesterolemia is an autosomal dominant disorder due to mutations in the low-density lipoprotein receptor gene, characterized by skin and tendon xanthomas, xanthelasma and premature arcus corneae. It is associated with an increased risk of premature coronary heart disease, which is further increased if there is co-existing diabetes mellitus. A 35-year-old female who developed cutaneous and tendon xanthomas since the age of 12 was diagnosed as having mixed primary familial hypercholesterolemia and secondary hyperlipidemia due to diabetes mellitus, and osteomyelitis. However, familial hypercholesterolemia remains seriously under-diagnosed, delaying treatment. Screening of first-degree relatives and extended family members plays an important role in early detection and treatment. ( J Intern Med Taiwan 2003;14:23-30 ) Key Words:Familial hypercholesterolemia, Xanthomatosis, Diabetes mellitus, LDL receptor, Osteomyelitis Introduction Familial hypercholesterolemia is a monogenic 1 , autosomal dominant 2 disorder due to mutations in the gene for the LDL receptor 3, characterized by xanthomas 4, xanthelasma and premature arcus corneae 5. Our report concerns a 35-year-old female patient who developed xanthomas since the age of 12 and was diagnosed as having mixed primary familial hypercholesterolemia and secondary hyperlipidemia due to diabetes mellitus, and osteomyelitis. The significance, characteristic features, diagnosis and treatment of familial hypercholesterolemia are discussed. -

Erdheim-Chester Disease and Eyes
Erdheim-Chester Disease and Eyes OMAR OZGUR, MD OCTOBER 10, 2015 9:30- 10:00AM OPHTHALMIC PLASTIC AND RECONSTRUCTIVE SURGERY AND ORBITAL ONCOLOGY FELLOW DEPARTMENT OF PLASTIC SURGERY THE UNIVERSITY OF TEXAS MD ANDERSON CANCER CENTER HOUSTON, TX, UNITED STATES Outline Anatomy and function of the eye and orbit Background and overview of ECD What can ECD do to the eye? DoubleDouble visionvision Pain Exophthalmos Overview of an eye exam General eye conditions that may also occur Droopy eyelids Dry eye syndrome Cataracts Refractive error Questions Please ask anytime! www.nasa.gov – Cat’s Eye Nebula Background anatomy https://c2.staticflickr.com/2/1363/542580866_940d2f9a02.jpg http://fiftylives.org/blog/wp- content/uploads/2012/08/Cornea.jpg Eye anatomy https://c2.staticflickr. com/2/1363/5425808 66_940d2f9a02.jpg http://www.retina-doctors.com/galleries/splash_patients/Eye%20anatomy%20-%20AN0003.jpg Optic nerves http://antranik.org/wp-content/uploads/2011/11/optic-nerve-lateral- geniculate-nucleus-of-thalamus-optic-radiation-visual-cortex.jpg?9873a6 http://www.vision-and-eye-health.com/images/GCA-AION.jpg Orbital anatomy http://www.leventefe.com.au/portfolio /mi-tec-medical-media-3/ http://msk- anatomy.blogspot.com/2014/12/ cranial-nerves-anatomy.html Eyelid structure http://eyestrain.sabhlokcity.com/2011/ 09/meibomian-gland-disease-mgd/ http://www.mastereyeassociates.com/blepharitis http://www.medindia.net/patients/patientinfo/gran ulated-eyelids.htm CT and MRI http://www.ijri.org/articles/2012/2 2/3/images/IndianJRadiolImaging_ -

Vaccinations for Adults with Chronic Liver Disease Or Infection
Vaccinations for Adults with Chronic Liver Disease or Infection This table shows which vaccinations you should have to protect your health if you have chronic hepatitis B or C infection or chronic liver disease (e.g., cirrhosis). Make sure you and your healthcare provider keep your vaccinations up to date. Vaccine Do you need it? Hepatitis A Yes! Your chronic liver disease or infection puts you at risk for serious complications if you get infected with the (HepA) hepatitis A virus. If you’ve never been vaccinated against hepatitis A, you need 2 doses of this vaccine, usually spaced 6–18 months apart. Hepatitis B Yes! If you already have chronic hepatitis B infection, you won’t need hepatitis B vaccine. However, if you have (HepB) hepatitis C or other causes of chronic liver disease, you do need hepatitis B vaccine. The vaccine is given in 2 or 3 doses, depending on the brand. Ask your healthcare provider if you need screening blood tests for hepatitis B. Hib (Haemophilus Maybe. Some adults with certain high-risk conditions, for example, lack of a functioning spleen, need vaccination influenzae type b) with Hib. Talk to your healthcare provider to find out if you need this vaccine. Human Yes! You should get this vaccine if you are age 26 years or younger. Adults age 27 through 45 may also be vacci- papillomavirus nated against HPV after a discussion with their healthcare provider. The vaccine is usually given in 3 doses over a (HPV) 6-month period. Influenza Yes! You need a dose every fall (or winter) for your protection and for the protection of others around you.