A Drug-Induced Cholestatic Pattern
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of Abnormal Liver Chemistries
ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries Paul Y. Kwo, MD, FACG, FAASLD1, Stanley M. Cohen, MD, FACG, FAASLD2, and Joseph K. Lim, MD, FACG, FAASLD3 1Division of Gastroenterology/Hepatology, Department of Medicine, Stanford University School of Medicine, Palo Alto, California, USA; 2Digestive Health Institute, University Hospitals Cleveland Medical Center and Division of Gastroenterology and Liver Disease, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA; 3Yale Viral Hepatitis Program, Yale University School of Medicine, New Haven, Connecticut, USA. Am J Gastroenterol 2017; 112:18–35; doi:10.1038/ajg.2016.517; published online 20 December 2016 Abstract Clinicians are required to assess abnormal liver chemistries on a daily basis. The most common liver chemistries ordered are serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase and bilirubin. These tests should be termed liver chemistries or liver tests. Hepatocellular injury is defined as disproportionate elevation of AST and ALT levels compared with alkaline phosphatase levels. Cholestatic injury is defined as disproportionate elevation of alkaline phosphatase level as compared with AST and ALT levels. The majority of bilirubin circulates as unconjugated bilirubin and an elevated conjugated bilirubin implies hepatocellular disease or cholestasis. Multiple studies have demonstrated that the presence of an elevated ALT has been associated with increased liver-related mortality. A true healthy normal ALT level ranges from 29 to 33 IU/l for males, 19 to 25 IU/l for females and levels above this should be assessed. The degree of elevation of ALT and or AST in the clinical setting helps guide the evaluation. -

Acute Liver Failure J G O’Grady
148 Postgrad Med J: first published as 10.1136/pgmj.2004.026005 on 4 March 2005. Downloaded from REVIEW Acute liver failure J G O’Grady ............................................................................................................................... Postgrad Med J 2005;81:148–154. doi: 10.1136/pgmj.2004.026005 Acute liver failure is a complex multisystemic illness that account for most cases, but a significant number of patients have no definable cause and are evolves quickly after a catastrophic insult to the liver classified as seronegative or of being of indeter- leading to the development of encephalopathy. The minate aetiology. Paracetamol is the commonest underlying aetiology and the pace of progression strongly cause in the UK and USA.2 Idiosyncratic reac- tions comprise another important group. influence the clinical course. The commonest causes are paracetamol, idiosyncratic drug reactions, hepatitis B, and Viral seronegative hepatitis. The optimal care is multidisciplinary ALF is an uncommon complication of viral and up to half of the cases receive liver transplants, with hepatitis, occurring in 0.2%–4% of cases depend- ing on the underlying aetiology.3 The risk is survival rates around 75%–90%. Artificial liver support lowest with hepatitis A, but it increases with the devices remain unproven in efficacy in acute liver failure. age at time of exposure. Hepatitis B can be associated with ALF through a number of ........................................................................... scenarios (table 2). The commonest are de novo infection and spontaneous surges in viral repli- cation, while the incidence of the delta virus cute liver failure (ALF) is a complex infection seems to be decreasing rapidly. multisystemic illness that evolves after a Vaccination should reduce the incidence of Acatastrophic insult to the liver manifesting hepatitis A and B, while antiviral drugs should in the development of a coagulopathy and ameliorate replication of hepatitis B. -

A Rare Hepatic Manifestation of Systemic Lupus Erythematosus
Cholestatic hepatitis in SLE Cholestatichepatitis:ararehepaticmanifestationof systemiclupuserythematosus WHChow,MSLam,WKKwan,WFNg Systemic lupus erythematosus is a multi-system inflammatory disease. The clinical manifestations are diverse. Hepatic manifestation is a rarely seen complication of systemic lupus erythematosus. We report a case of complication of systemic lupus erythematosus presenting as cholestatic hepatitis in a 56-year- old Chinese woman. The cholestatic hepatitis progressed as part of the lupus activity and responded to steroid therapy. HKMJ 1997;3:331-4 Key words: Hepatitis; Cholestasis; Lupus erythematosus, systemic; Liver Introduction of body weight and had had a poor appetite. She was a non-drinker and had no long term drug history. Systemic lupus erythematosus (SLE) is a multi- system inflammatory disease associated with the A general examination showed her to be jaundiced, development of auto-antibodies to a variety of self- pale, and dyspnoeic with an elevated body tempera- antigens. The clinical manifestations of SLE are ture of 38.2°C. Chest examination demonstrated coarse diverse. In 1982, the American Rheumatism Associa- crackles heard over both lung fields. Other parts of tion (ARA) published revised criteria for the classifi- the examination were unremarkable. There was 2+ cation of SLE.1 For a diagnosis of SLE, individuals proteinuria in the mid-stream urine but the culture should have four or more of the following features: for organisms was negative. Investigations revealed a malar rash, discoid rash, photosensitivity, oral ulcers, normochromic, normocytic anaemia (haemoglobin 9.4 non-erosive arthritis, pleuritis or pericarditis, renal g/dL [normal range, 11.5-15.5 g/dL]) with normal disorder, seizures or psychosis, haematological white cell and differential counts. -

Primary Biliary Cholangitis: a Brief Overview Justin S
REVIEW Primary Biliary Cholangitis: A Brief Overview Justin S. Louie,* Sirisha Grandhe,* Karen Matsukuma,† and Christopher L. Bowlus* Primary biliary cholangitis (PBC), previously referred to supported by the higher concordance of PBC in monozy- as primary biliary cirrhosis, is the most common chronic gotic compared with dizygotic twins.4 In addition, certain cholestatic autoimmune disease affecting adults in the human leukocyte antigen haplotypes have been associ- United States.1 It is characterized by a hallmark serologic ated with PBC, as well as variants at loci along the inter- signature, antimitochondrial antibody (AMA), and specific leukin-12 (IL-12) immunoregulatory pathway (IL-12A and bile duct pathology with progressive intrahepatic duct de- IL-12RB2 loci).5 struction leading to cholestasis. PBC is potentially fatal and can have both intrahepatic and extrahepatic complications. PATHOGENESIS EPIDEMIOLOGY The primary disease mechanism in PBC is thought to be T cell lymphocyte–mediated injury against intralobu- PBC affects all races and ethnicities; however, it is best lar biliary epithelial cells. This causes progressive destruc- studied in the Caucasian population. The condition pre- tion and eventual disappearance of the intralobular bile dominantly affects women older than 40 years, with a ducts. Molecular mimicry has been proposed as the ini- female/male ratio of 9:1.2 Although the incidence of PBC tiating event in the loss of tolerance primarily to mito- appears to be stable, the overall prevalence of the disease chondrial pyruvate dehydrogenase complex, E2, during is increasing.3 An individual’s genetic susceptibility, epige- which exogenous antigens evoke an immune response netic factors, and certain environmental triggers seem to that recognizes an endogenous (self) antigen inciting an play important roles. -
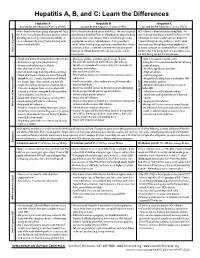
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Progress Report Cholestasis and Lesions of the Biliary Tract in Chronic Pancreatitis
Gut: first published as 10.1136/gut.19.9.851 on 1 September 1978. Downloaded from Gut, 1978, 19, 851-857 Progress report Cholestasis and lesions of the biliary tract in chronic pancreatitis The occurrence of jaundice in the course of chronic pancreatitis has been recognised since the 19th century" 2. But in the early papers it is uncertain whether the cases were due to acute, acute relapsing, or to chronic pan- creatitis, or even to pancreatic cancer associated with pancreatitis or benign ampullary stenosis. With the introduction of endoscopic retrograde cholangiopancreato- graphy (ERCP), there has been a renewed interest in the biliary complica- tions of chronic pancreatitis (CP). However, papers published recently by endoscopists have generally neglected the cholangiographic aspect of the lesions and are less precise and less well documented than papers published just after the second world war, following the introduction of manometric cholangiography3-5. Furthermore, the description of obstructive jaundice due to chronic pancreatitis, classical 20 years ago, seems to have been forgotten until the recent papers. Radiological aspects of bile ducts in chronic pancreatitis http://gut.bmj.com/ If one limits the subject to primary diseases of the pancreas, particularly chronic calcifying pancreatitis (CCP)6, excluding chronic pancreatitis secondary to benign ampullary stenosis7, cancer obstructing the main pancreatic duct8 9 and acute relapsing pancreatitis secondary to gallstones'0 radiological aspect of the main bile duct" is type I the most.common on September 25, 2021 by guest. Protected copyright. choledocus (Figure). This description has been repeatedly confirmed'2"13. It is a long stenosis of the intra- or retropancreatic part of the main bile duct. -

Diagnosis and Management of Primary Biliary Cholangitis Ticle
REVIEW ArtICLE 1 see related editorial on page x Diagnosis and Management of Primary Biliary Cholangitis TICLE R Zobair M. Younossi, MD, MPH, FACG, AGAF, FAASLD1, David Bernstein, MD, FAASLD, FACG, AGAF, FACP2, Mitchell L. Shifman, MD3, Paul Kwo, MD4, W. Ray Kim, MD5, Kris V. Kowdley, MD6 and Ira M. Jacobson, MD7 Primary biliary cholangitis (PBC) is a chronic, cholestatic, autoimmune disease with a variable progressive course. PBC can cause debilitating symptoms including fatigue and pruritus and, if left untreated, is associated with a high risk of cirrhosis and related complications, liver failure, and death. Recent changes to the PBC landscape include a REVIEW A name change, updated guidelines for diagnosis and treatment as well as new treatment options that have recently become available. Practicing clinicians face many unanswered questions when managing PBC. To assist these healthcare providers in managing patients with PBC, the American College of Gastroenterology (ACG) Institute for Clinical Research & Education, in collaboration with the Chronic Liver Disease Foundation (CLDF), organized a panel of experts to evaluate and summarize the most current and relevant peer-reviewed literature regarding PBC. This, combined with the extensive experience and clinical expertise of this expert panel, led to the formation of this clinical guidance on the diagnosis and management of PBC. Am J Gastroenterol https://doi.org/10.1038/s41395-018-0390-3 INTRODUCTION addition, diagnosis and treatment guidelines are changing and a Primary biliary cholangitis (PBC) is a chronic, cholestatic, auto- number of guidelines have been updated [4, 5]. immune disease with a progressive course that may extend over Because of important changes in the PBC landscape, and a num- many decades. -

An Update on the Management of Cholestatic Liver Diseases
Clinical Medicine 2020 Vol 20, No 5: 513–6 CME: HEPATOLOGY An update on the management of cholestatic liver diseases Authors: Gautham AppannaA and Yiannis KallisB Cholestatic liver diseases are a challenging spectrum of autoantibodies though may be performed in cases of diagnostic conditions arising from damage to bile ducts, leading to doubt, suspected overlap syndrome or co-existent liver pathology build-up of bile acids and inflammatory processes that cause such as fatty liver. injury to cholangiocytes and hepatocytes. Primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) are ABSTRACT the two most common cholestatic disorders. In this review we Key points detail the latest guidelines for the diagnosis and management of patients with these two conditions. Ursodeoxycholic acid can favourably alter the natural history of primary biliary cholangitis in a majority of patients, given at the appropriate dose of 13–15 mg/kg/day. Primary biliary cholangitis Primary biliary cholangitis (PBC) is a chronic autoimmune liver Risk stratification is of paramount importance in the disorder characterised by immune-mediated destruction of management of primary biliary cholangitis to identify epithelial cells lining the intrahepatic bile ducts, resulting in patients with suboptimal response to ursodeoxycholic acid persistent cholestasis and, in some patients, a progression to and poorer long-term prognosis. Alkaline phosphatase cirrhosis if left untreated. The exact mechanisms remain unclear >1.67 times the upper limit of normal and a bilirubin above but are most likely a result of exposure to environmental factors in the normal range indicate high-risk disease and suboptimal a genetically susceptible individual.1 The majority of patients are treatment response. -

Guideline for the Evaluation of Cholestatic Jaundice
CLINICAL GUIDELINES Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition ÃRima Fawaz, yUlrich Baumann, zUdeme Ekong, §Bjo¨rn Fischler, jjNedim Hadzic, ôCara L. Mack, #Vale´rie A. McLin, ÃÃJean P. Molleston, yyEzequiel Neimark, zzVicky L. Ng, and §§Saul J. Karpen ABSTRACT Cholestatic jaundice in infancy affects approximately 1 in every 2500 term PREAMBLE infants and is infrequently recognized by primary providers in the setting of holestatic jaundice in infancy is an uncommon but poten- physiologic jaundice. Cholestatic jaundice is always pathologic and indicates tially serious problem that indicates hepatobiliary dysfunc- hepatobiliary dysfunction. Early detection by the primary care physician and tion.C Early detection of cholestatic jaundice by the primary care timely referrals to the pediatric gastroenterologist/hepatologist are important physician and timely, accurate diagnosis by the pediatric gastro- contributors to optimal treatment and prognosis. The most common causes of enterologist are important for successful treatment and an optimal cholestatic jaundice in the first months of life are biliary atresia (25%–40%) prognosis. The Cholestasis Guideline Committee consisted of 11 followed by an expanding list of monogenic disorders (25%), along with many members of 2 professional societies: the North American Society unknown or multifactorial (eg, parenteral nutrition-related) causes, each of for Gastroenterology, Hepatology and Nutrition, and the European which may have time-sensitive and distinct treatment plans. Thus, these Society for Gastroenterology, Hepatology and Nutrition. This guidelines can have an essential role for the evaluation of neonatal cholestasis committee has responded to a need in pediatrics and developed to optimize care. -

Sudden (Acute) Liver Failure
Customer Name, Street Address, City, State, Zip code Phone number, Alt. phone number, Fax number, e-mail address, web site Sudden (Acute) Liver Failure Basics OVERVIEW • Sudden (acute) damage to the liver tissue that is so severe that the liver is unable to function properly and meet the needs of the body • Sudden (acute) loss of more than 75% of functional liver tissue; occurs primarily because of sudden (acute), massive death of liver tissue (known as “hepatic necrosis”), which has a catastrophic effect on multiple organs in a previously healthy pet; may rapidly lead to death • The liver is the largest gland in the body; it has many functions, including production of bile (a fluid substance involved in digestion of fats); production of albumin (a protein in the plasma of the blood); and detoxification of drugs and other chemicals (such as ammonia) in the body SIGNALMENT/DESCRIPTION OF PET Species • Dogs • Cats • More common in dogs than in cats Breed Predilections • Breeds that appear to have increased likelihood of having long-term inflammation of the liver (known as “chronic hepatitis”) as compared to other breeds may have higher risk of sudden (acute) liver failure SIGNS/OBSERVED CHANGES IN THE PET • Sudden (acute) onset • Sluggishness (lethargy) • Decreased appetite (known as “inappetence”) • Vomiting • Small intestinal diarrhea—may be bloody • Increased thirst (known as “polydipsia”) and increased urination (known as “polyuria”) • Enlargement of the liver (known as “hepatomegaly”), with tenderness of the liver on feeling the abdomen -

COVID-19 and Liver Cirrhosis Important Information for Patients and Their Families
COVID-19 and Liver Cirrhosis Important Information for Patients and Their Families The American Association for the Study of Liver Diseases (AASLD) is committed to helping you understand coronavirus disease 2019 (COVID-19) infection and prevention in people with liver cirrhosis. What We Know Our understanding of COVID-19 in people with liver cirrhosis is evolving. When making decisions related to COVID-19 infections or prevention, having up-to-date information is critical. • Symptoms of COVID-19 infection include any of the following: fever, chills, drowsiness, cough, congestion or runny nose, difficulty breathing, fatigue, body aches, headache, sore throat, abdominal pain, nausea, vomiting, diarrhea, and loss of sense of taste or smell. • People with underlying cirrhosis of the liver are at a higher risk of developing severe COVID-19 illness and/or more problems from their existing liver disease if they get a COVID-19 infection, with prolonged hospitalization and increased mortality. These patients need to take careful precautions to avoid COVID-19 infection. COVID-19 may affect the processes and procedures for screening, diagnosis, and treatment of liver cirrhosis. • Cirrhosis, or scarring of the liver, can be caused by many chronic liver diseases, including viral hepatitis, as well as excessive alcohol intake, obesity, diabetes, diseases of the bile ducts, and a variety of toxic, metabolic, or other inherited diseases. • Most people with liver disease are asymptomatic. Complications, such as yellowing of the skin and eyes from jaundice, internal bleeding (varices), mental confusion (hepatic encephalopathy), and/or swollen belly from ascites, may take years to develop, so patients are often unaware of the severity of their condition and the slow, progressive damage. -

Diagnosis and Management of Primary Sclerosing Cholangitis
AASLD PRACTICE GUIDELINES Diagnosis and Management of Primary Sclerosing Cholangitis Roger Chapman,1 Johan Fevery,2 Anthony Kalloo,3 David M. Nagorney,4 Kirsten Muri Boberg,5 Benjamin Shneider,6 and Gregory J. Gores7 Preamble classification used by the Grading of Recommendation This guideline has been approved by the American Asso- Assessment, Development, and Evaluation (GRADE) ciation for the Study of Liver Diseases and represents the workgroup with minor modifications (Table 1).3 The position of the Association. These recommendations pro- strength of recommendations in the GRADE system are vide a data-supported approach. They are based on the classified as strong (class 1) or weak (class 2). The quality following: (1) formal review and analysis of the recently- of evidence supporting strong or weak recommendations published world literature on the topic (Medline search); is designated by one of three levels: high (level A), mod- (2) American College of Physicians Manual for Assessing erate (level B), or low-quality (level C). Health Practices and Designing Practice Guidelines1; (3) guideline policies, including the AASLD Policy on the Definition and Diagnosis Development and Use of Practice Guidelines and the Definitions. Primary sclerosing cholangitis (PSC) is a American Gastroenterological Association Policy State- chronic, cholestatic liver disease characterized by inflam- ment on Guidelines2; and (4) the experience of the au- mation and fibrosis of both intrahepatic and extrahepatic thors in the specified topic. bile ducts,4 leading to the formation of multifocal bile Intended for use by physicians, these recommenda- duct strictures. PSC is likely an immune mediated, pro- tions suggest preferred approaches to the diagnostic, ther- gressive disorder that eventually develops into cirrhosis, apeutic and preventative aspects of care.