Definition and Treatment of Liver Failure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Evaluation of Abnormal Liver Chemistries
ACG Clinical Guideline: Evaluation of Abnormal Liver Chemistries Paul Y. Kwo, MD, FACG, FAASLD1, Stanley M. Cohen, MD, FACG, FAASLD2, and Joseph K. Lim, MD, FACG, FAASLD3 1Division of Gastroenterology/Hepatology, Department of Medicine, Stanford University School of Medicine, Palo Alto, California, USA; 2Digestive Health Institute, University Hospitals Cleveland Medical Center and Division of Gastroenterology and Liver Disease, Department of Medicine, Case Western Reserve University School of Medicine, Cleveland, Ohio, USA; 3Yale Viral Hepatitis Program, Yale University School of Medicine, New Haven, Connecticut, USA. Am J Gastroenterol 2017; 112:18–35; doi:10.1038/ajg.2016.517; published online 20 December 2016 Abstract Clinicians are required to assess abnormal liver chemistries on a daily basis. The most common liver chemistries ordered are serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase and bilirubin. These tests should be termed liver chemistries or liver tests. Hepatocellular injury is defined as disproportionate elevation of AST and ALT levels compared with alkaline phosphatase levels. Cholestatic injury is defined as disproportionate elevation of alkaline phosphatase level as compared with AST and ALT levels. The majority of bilirubin circulates as unconjugated bilirubin and an elevated conjugated bilirubin implies hepatocellular disease or cholestasis. Multiple studies have demonstrated that the presence of an elevated ALT has been associated with increased liver-related mortality. A true healthy normal ALT level ranges from 29 to 33 IU/l for males, 19 to 25 IU/l for females and levels above this should be assessed. The degree of elevation of ALT and or AST in the clinical setting helps guide the evaluation. -

A Drug-Induced Cholestatic Pattern
Review articles Hepatotoxicity: A Drug-Induced Cholestatic Pattern Laura Morales M.,1 Natalia Vélez L.,1 Octavio Germán Muñoz M., MD.2 1 Medical Student in the Faculty of Medicine and Abstract the Gastrohepatology Group at the Universidad de Antioquia in Medellín, Colombia Although drug induced liver disease is a rare condition, it explains 40% to 50% of all cases of acute liver 2 Internist and Hepatologist at the Hospital Pablo failure. In 20% to 40% of the cases, the pattern is cholestatic and is caused by inhibition of the transporters Tobon Uribe and in the Gastrohepatology Group at that regulate bile synthesis. This reduction in activity is directly or indirectly mediated by drugs and their me- the Universidad de Antioquia in Medellín, Colombia tabolites and/or by genetic polymorphisms and other risk factors of the patient. Its manifestations range from ......................................... biochemical alterations in the absence of symptoms to acute liver failure and chronic liver damage. Received: 30-01-15 Although there is no absolute test or marker for diagnosis of this disease, scales and algorithms have Accepted: 26-01-16 been developed to assess the likelihood of cholestatic drug induced liver disease. Other types of evidence are not routinely used because of their complexity and cost. Diagnosis is primarily based on exclusion using circumstantial evidence. Cholestatic drug induced liver disease has better overall survival rates than other patters, but there are higher risks of developing chronic liver disease. In most cases, the patient’s condition improves when the drug responsible for the damage is removed. Hemodialysis and transplantation should be considered only for selected cases. -

Acute Liver Failure J G O’Grady
148 Postgrad Med J: first published as 10.1136/pgmj.2004.026005 on 4 March 2005. Downloaded from REVIEW Acute liver failure J G O’Grady ............................................................................................................................... Postgrad Med J 2005;81:148–154. doi: 10.1136/pgmj.2004.026005 Acute liver failure is a complex multisystemic illness that account for most cases, but a significant number of patients have no definable cause and are evolves quickly after a catastrophic insult to the liver classified as seronegative or of being of indeter- leading to the development of encephalopathy. The minate aetiology. Paracetamol is the commonest underlying aetiology and the pace of progression strongly cause in the UK and USA.2 Idiosyncratic reac- tions comprise another important group. influence the clinical course. The commonest causes are paracetamol, idiosyncratic drug reactions, hepatitis B, and Viral seronegative hepatitis. The optimal care is multidisciplinary ALF is an uncommon complication of viral and up to half of the cases receive liver transplants, with hepatitis, occurring in 0.2%–4% of cases depend- ing on the underlying aetiology.3 The risk is survival rates around 75%–90%. Artificial liver support lowest with hepatitis A, but it increases with the devices remain unproven in efficacy in acute liver failure. age at time of exposure. Hepatitis B can be associated with ALF through a number of ........................................................................... scenarios (table 2). The commonest are de novo infection and spontaneous surges in viral repli- cation, while the incidence of the delta virus cute liver failure (ALF) is a complex infection seems to be decreasing rapidly. multisystemic illness that evolves after a Vaccination should reduce the incidence of Acatastrophic insult to the liver manifesting hepatitis A and B, while antiviral drugs should in the development of a coagulopathy and ameliorate replication of hepatitis B. -

Management of Autoimmune Liver Diseases After Liver Transplantation
Review Management of Autoimmune Liver Diseases after Liver Transplantation Romelia Barba Bernal 1,† , Esli Medina-Morales 1,† , Daniela Goyes 2 , Vilas Patwardhan 1 and Alan Bonder 1,* 1 Division of Gastroenterology and Hepatology, Beth Israel Deaconess Medical Center, Boston, MA 02215, USA; [email protected] (R.B.B.); [email protected] (E.M.-M.); [email protected] (V.P.) 2 Department of Medicine, Loyola Medicine—MacNeal Hospital, Berwyn, IL 60402, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-617-632-1070 † These authors contributed equally to this project. Abstract: Autoimmune liver diseases are characterized by immune-mediated inflammation and even- tual destruction of the hepatocytes and the biliary epithelial cells. They can progress to irreversible liver damage requiring liver transplantation. The post-liver transplant goals of treatment include improving the recipient’s survival, preventing liver graft-failure, and decreasing the recurrence of the disease. The keystone in post-liver transplant management for autoimmune liver diseases relies on identifying which would be the most appropriate immunosuppressive maintenance therapy. The combination of a steroid and a calcineurin inhibitor is the current immunosuppressive regimen of choice for autoimmune hepatitis. A gradual withdrawal of glucocorticoids is also recommended. Citation: Barba Bernal, R.; On the other hand, ursodeoxycholic acid should be initiated soon after liver transplant to prevent Medina-Morales, E.; Goyes, D.; recurrence and improve graft and patient survival in primary biliary cholangitis recipients. Unlike the Patwardhan, V.; Bonder, A. Management of Autoimmune Liver previously mentioned autoimmune diseases, there are not immunosuppressive or disease-modifying Diseases after Liver Transplantation. -

Spontaneous Bacterial Peritonitis: Recent Guidelines and Beyond R Wiest,1 a Krag,2 a Gerbes3
Downloaded from gut.bmj.com on May 31, 2012 - Published by group.bmj.com Recent advances in clinical practice Spontaneous bacterial peritonitis: recent guidelines and beyond R Wiest,1 A Krag,2 A Gerbes3 1Department for visceral surgery INTRODUCTION prognosis in various cohorts of patients with SBP and medicine, University Spontaneous bacterial peritonitis (SBP) is the most are summarised in figure 1 and include age,16 20 Hospital Bern, Switzerland 18 20 23 16 18 2 frequent and life-threatening infection in patients Child score, intensive care, nosocomial Department of 18 24 25 Gastroenterology, Hvidovre with liver cirrhosis requiring prompt recognition origin, hepatic encephalopathy, elevated 26 University Hospital, and treatment. It is defined by the presence of >250 serum creatinine and bilirubin, lack of infection Copenhagen, Denmark polymorphonuclear cells (PMN)/mm3 in ascites in resolution/need to escalate treatment and culture 3 e Klinikum of the University of the absence of an intra-abdominal source of infec- positivity27 29 as well as the presence of bacter- Munich, Munich, Germany tion or malignancy. In this review we discuss the aemia30 and CARD15/NOD2 variants as a genetic fl 31 Correspondence to current opinions re ected by recent guidelines risk factor. It is important to stress in this context Professor Dr Reiner Wiest, (American Association for the Study of Liver that the only factors that are modifiable in this Department for visceral surgery Diseases, European Association for the Study of the scenario are timely diagnosis and effective first-line and medicine, University Liver, Deutsche Gesellschaft für Verdauungs- und treatment. Hospital Bern, 3010 Bern, 1e4 Switzerland; Stoffwechselkrankheiten), with particular focus [email protected] on controversial issues as well as open questions Bacterial translocation (BT) and pathophysiology that need to be addressed in the future. -

Primary Biliary Cholangitis: a Brief Overview Justin S
REVIEW Primary Biliary Cholangitis: A Brief Overview Justin S. Louie,* Sirisha Grandhe,* Karen Matsukuma,† and Christopher L. Bowlus* Primary biliary cholangitis (PBC), previously referred to supported by the higher concordance of PBC in monozy- as primary biliary cirrhosis, is the most common chronic gotic compared with dizygotic twins.4 In addition, certain cholestatic autoimmune disease affecting adults in the human leukocyte antigen haplotypes have been associ- United States.1 It is characterized by a hallmark serologic ated with PBC, as well as variants at loci along the inter- signature, antimitochondrial antibody (AMA), and specific leukin-12 (IL-12) immunoregulatory pathway (IL-12A and bile duct pathology with progressive intrahepatic duct de- IL-12RB2 loci).5 struction leading to cholestasis. PBC is potentially fatal and can have both intrahepatic and extrahepatic complications. PATHOGENESIS EPIDEMIOLOGY The primary disease mechanism in PBC is thought to be T cell lymphocyte–mediated injury against intralobu- PBC affects all races and ethnicities; however, it is best lar biliary epithelial cells. This causes progressive destruc- studied in the Caucasian population. The condition pre- tion and eventual disappearance of the intralobular bile dominantly affects women older than 40 years, with a ducts. Molecular mimicry has been proposed as the ini- female/male ratio of 9:1.2 Although the incidence of PBC tiating event in the loss of tolerance primarily to mito- appears to be stable, the overall prevalence of the disease chondrial pyruvate dehydrogenase complex, E2, during is increasing.3 An individual’s genetic susceptibility, epige- which exogenous antigens evoke an immune response netic factors, and certain environmental triggers seem to that recognizes an endogenous (self) antigen inciting an play important roles. -
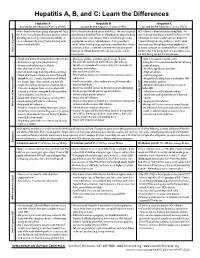
Hepatitis A, B, and C: Learn the Differences
Hepatitis A, B, and C: Learn the Differences Hepatitis A Hepatitis B Hepatitis C caused by the hepatitis A virus (HAV) caused by the hepatitis B virus (HBV) caused by the hepatitis C virus (HCV) HAV is found in the feces (poop) of people with hepa- HBV is found in blood and certain body fluids. The virus is spread HCV is found in blood and certain body fluids. The titis A and is usually spread by close personal contact when blood or body fluid from an infected person enters the body virus is spread when blood or body fluid from an HCV- (including sex or living in the same household). It of a person who is not immune. HBV is spread through having infected person enters another person’s body. HCV can also be spread by eating food or drinking water unprotected sex with an infected person, sharing needles or is spread through sharing needles or “works” when contaminated with HAV. “works” when shooting drugs, exposure to needlesticks or sharps shooting drugs, through exposure to needlesticks on the job, or from an infected mother to her baby during birth. or sharps on the job, or sometimes from an infected How is it spread? Exposure to infected blood in ANY situation can be a risk for mother to her baby during birth. It is possible to trans- transmission. mit HCV during sex, but it is not common. • People who wish to be protected from HAV infection • All infants, children, and teens ages 0 through 18 years There is no vaccine to prevent HCV. -

Diagnosis and Management of Primary Biliary Cholangitis Ticle
REVIEW ArtICLE 1 see related editorial on page x Diagnosis and Management of Primary Biliary Cholangitis TICLE R Zobair M. Younossi, MD, MPH, FACG, AGAF, FAASLD1, David Bernstein, MD, FAASLD, FACG, AGAF, FACP2, Mitchell L. Shifman, MD3, Paul Kwo, MD4, W. Ray Kim, MD5, Kris V. Kowdley, MD6 and Ira M. Jacobson, MD7 Primary biliary cholangitis (PBC) is a chronic, cholestatic, autoimmune disease with a variable progressive course. PBC can cause debilitating symptoms including fatigue and pruritus and, if left untreated, is associated with a high risk of cirrhosis and related complications, liver failure, and death. Recent changes to the PBC landscape include a REVIEW A name change, updated guidelines for diagnosis and treatment as well as new treatment options that have recently become available. Practicing clinicians face many unanswered questions when managing PBC. To assist these healthcare providers in managing patients with PBC, the American College of Gastroenterology (ACG) Institute for Clinical Research & Education, in collaboration with the Chronic Liver Disease Foundation (CLDF), organized a panel of experts to evaluate and summarize the most current and relevant peer-reviewed literature regarding PBC. This, combined with the extensive experience and clinical expertise of this expert panel, led to the formation of this clinical guidance on the diagnosis and management of PBC. Am J Gastroenterol https://doi.org/10.1038/s41395-018-0390-3 INTRODUCTION addition, diagnosis and treatment guidelines are changing and a Primary biliary cholangitis (PBC) is a chronic, cholestatic, auto- number of guidelines have been updated [4, 5]. immune disease with a progressive course that may extend over Because of important changes in the PBC landscape, and a num- many decades. -

Non-Alcoholic Fatty Liver Disease
Non-alcoholic fatty liver disease Description Non-alcoholic fatty liver disease (NAFLD) is a buildup of excessive fat in the liver that can lead to liver damage resembling the damage caused by alcohol abuse, but that occurs in people who do not drink heavily. The liver is a part of the digestive system that helps break down food, store energy, and remove waste products, including toxins. The liver normally contains some fat; an individual is considered to have a fatty liver (hepatic steatosis) if the liver contains more than 5 to 10 percent fat. The fat deposits in the liver associated with NAFLD usually cause no symptoms, although they may cause increased levels of liver enzymes that are detected in routine blood tests. Some affected individuals have abdominal pain or fatigue. During a physical examination, the liver may be found to be slightly enlarged. Between 7 and 30 percent of people with NAFLD develop inflammation of the liver (non- alcoholic steatohepatitis, also known as NASH), leading to liver damage. Minor damage to the liver can be repaired by the body. However, severe or long-term damage can lead to the replacement of normal liver tissue with scar tissue (fibrosis), resulting in irreversible liver disease (cirrhosis) that causes the liver to stop working properly. Signs and symptoms of cirrhosis, which get worse as fibrosis affects more of the liver, include fatigue, weakness, loss of appetite, weight loss, nausea, swelling (edema), and yellowing of the skin and whites of the eyes (jaundice). Scarring in the vein that carries blood into the liver from the other digestive organs (the portal vein) can lead to increased pressure in that blood vessel (portal hypertension), resulting in swollen blood vessels (varices) within the digestive system. -

Managing Liver Failure D a Kelly
660 Postgrad Med J: first published as 10.1136/pmj.78.925.660 on 1 November 2002. Downloaded from BEST PRACTICE Managing liver failure D A Kelly ............................................................................................................................. Postgrad Med J 2002;78:660–667 Liver disease is rare in childhood, but important new Clinical presentation developments have altered the natural history and The clinical presentation depends on the aeti- ology of acute liver failure but the presentation outcome. It is important that clinicians are aware of may either be acute, within days, or prolonged to these diseases and their management. Acute liver failure 10 weeks if due to metabolic liver disease. The is most often due to viral hepatitis, paracetamol extent of jaundice and encephalopathy is variable in the early stages, but all children have coagu- overdose, or inherited metabolic liver disease. The lopathy. Encephalopathy is particularly difficult to clinical presentation includes jaundice, coagulopathy, diagnose in neonates. Vomiting and poor feeding and encephalopathy. Early diagnosis is necessary to are early signs, while irritability and reversal of day/night sleep patterns indicates more estab- prevent complications such as cerebral oedema, lished hepatic encephalopathy. In older children gastrointestinal bleeding, and renal failure. Early encephalopathy may present with aggressive supportive management, in particular intravenous behaviour or convulsions. N-acetylcysteine, may be effective but liver Neonatal acute liver failure transplantation is usually the definitive treatment and The commonest cause of acute liver failure in thus early referral to a specialist unit for liver neonates is septicaemia secondary to Escherichia coli, Staphylococcus aureus, or herpes simplex (box 1 transplantation is mandatory. Chronic liver failure may and table 1). -

Vaccinations for Adults with Chronic Liver Disease Or Infection
Vaccinations for Adults with Chronic Liver Disease or Infection This table shows which vaccinations you should have to protect your health if you have chronic hepatitis B or C infection or chronic liver disease (e.g., cirrhosis). Make sure you and your healthcare provider keep your vaccinations up to date. Vaccine Do you need it? Hepatitis A Yes! Your chronic liver disease or infection puts you at risk for serious complications if you get infected with the (HepA) hepatitis A virus. If you’ve never been vaccinated against hepatitis A, you need 2 doses of this vaccine, usually spaced 6–18 months apart. Hepatitis B Yes! If you already have chronic hepatitis B infection, you won’t need hepatitis B vaccine. However, if you have (HepB) hepatitis C or other causes of chronic liver disease, you do need hepatitis B vaccine. The vaccine is given in 2 or 3 doses, depending on the brand. Ask your healthcare provider if you need screening blood tests for hepatitis B. Hib (Haemophilus Maybe. Some adults with certain high-risk conditions, for example, lack of a functioning spleen, need vaccination influenzae type b) with Hib. Talk to your healthcare provider to find out if you need this vaccine. Human Yes! You should get this vaccine if you are age 26 years or younger. Adults age 27 through 45 may also be vacci- papillomavirus nated against HPV after a discussion with their healthcare provider. The vaccine is usually given in 3 doses over a (HPV) 6-month period. Influenza Yes! You need a dose every fall (or winter) for your protection and for the protection of others around you. -

Acute Liver Failure
Acute Liver Failure ••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••••• When liver failure occurs in a previou ly healthy individual, it onset is often o rapid that it take experienced physicians by WILLIAM M. LEE surprise. Terms such a hyperacute or fulminant hepatic necrosis have been u ed to emphasize the rapidity and everity of the JULIE POLSON condition. The onset of acute liver failure is heralded by an altered mental state, often with agitation and confusion that may proceed to coma, and by evidence of coagulopathy [1 - 4]. The diagno is of acute liver failure require that these symptom develop in a patient without preexisting chronic liver disease. Although not all observers agree on precise definitions, it is generally accepted that onset of liver-related illness (usually, but not always, accompanied by jaundice) les than 8 weeks before patient presentation with altered mentation and coagulopathy fits the designation of acute liver failure. Development of these signs within a week of onset of liver disease and as much as 6 months from the first sign of illness is sometimes referred to as hyperacute and ubacute liver failure, respectively. An immediate challenge in managing sud1 patients is deter mination of the cause of acute liver failure in each case. Wherea certain factors, such a extent of encephalopathy and development of various complications, have an impact on outcome, the underlying etiology best determines the likeli hood of spontaneous survival or the need for urgent liver trans plantation [3,4]. The primary cause of acute liver failure vary in prevalence in different region of the world, and include drug toxicity and viral infections; there i also a large category of cases in which the pecific etiology cannot be determined.