Common Homozygosity for Predicted Loss-Of-Function Variants Reveals Both Redundant and Advantageous Effects of Dispensable Human Genes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

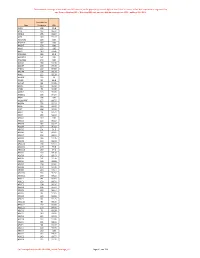
Onderstaande Coverage Is Berekend Over 1000 Exomen, Welke Geprept Zijn Met De Agilent Sureselect XT Exome V6 Kit
Onderstaande coverage is berekend over 1000 exomen, welke geprept zijn met de Agilent SureSelect XT exome v6 kit. Het sequencen is uitgevoerd op een Illumina NextSeq500 met een gemiddelde coverage van 100X , dekking 20x >95% over het gehele exoom. Gemiddelde Gen Coverage 20x A1BG 124 89,46 A1CF 123 98,03 A2ML1 120 98,19 A2M 114 95,38 A3GALT2 123 97,65 A4GALT 222 98,21 A4GNT 153 98,21 AAAS 147 98,21 AACS 139 98,20 AADACL2 118 97,73 AADACL3 139 98,21 AADACL4 156 98,21 AADAC 118 98,05 AADAT 107 89,53 AAED1 67 83,20 AAGAB 106 97,95 AAK1 115 97,79 AAMDC 101 88,39 AAMP 117 98,14 AANAT 124 98,16 AAR2 103 76,39 AARD 82 98,61 AARS2 133 98,12 AARSD1 105 84,15 AARS 123 98,17 AASDHPPT 127 97,08 AASDH 98 97,40 AASS 102 97,35 AATF 139 97,98 AATK 115 96,20 ABAT 111 94,77 ABCA1 124 97,79 ABCA2 152 97,17 ABCA3 129 98,12 ABCA4 126 98,14 ABCA5 59 88,83 ABCA6 88 95,52 ABCA7 163 98,07 ABCA8 102 96,10 ABCA9 109 97,71 ABCA10 74 85,59 ABCA12 116 97,77 ABCA13 129 96,43 ABCB1 114 97,45 ABCB4 96 96,93 ABCB5 105 97,75 ABCB6 140 98,21 ABCB7 126 99,13 ABCB8 140 98,05 ABCB9 139 98,13 ABCB10 85 89,17 ABCB11 118 97,74 ABCC1 123 92,71 ABCC2 127 98,16 ABCC3 147 97,94 ABCC4 112 96,52 ABCC5 121 92,63 ABCC6 115 91,98 ABCC8 129 98,17 ABCC9 108 97,76 ABCC10 144 97,99 ABCC11 126 98,16 ABCC12 134 98,20 ABCD1 113 72,00 ABCD2 96 97,46 ABCD3 90 91,11 ABCD4 118 98,16 ABCE1 63 86,86 ABCF1 116 98,04 Pagina 1 van 295 Onderstaande coverage is berekend over 1000 exomen, welke geprept zijn met de Agilent SureSelect XT exome v6 kit. -

An Evolutionary Based Strategy for Predicting Rational Mutations in G Protein-Coupled Receptors
Ecology and Evolutionary Biology 2021; 6(3): 53-77 http://www.sciencepublishinggroup.com/j/eeb doi: 10.11648/j.eeb.20210603.11 ISSN: 2575-3789 (Print); ISSN: 2575-3762 (Online) An Evolutionary Based Strategy for Predicting Rational Mutations in G Protein-Coupled Receptors Miguel Angel Fuertes*, Carlos Alonso Department of Microbiology, Centre for Molecular Biology “Severo Ochoa”, Spanish National Research Council and Autonomous University, Madrid, Spain Email address: *Corresponding author To cite this article: Miguel Angel Fuertes, Carlos Alonso. An Evolutionary Based Strategy for Predicting Rational Mutations in G Protein-Coupled Receptors. Ecology and Evolutionary Biology. Vol. 6, No. 3, 2021, pp. 53-77. doi: 10.11648/j.eeb.20210603.11 Received: April 24, 2021; Accepted: May 11, 2021; Published: July 13, 2021 Abstract: Capturing conserved patterns in genes and proteins is important for inferring phenotype prediction and evolutionary analysis. The study is focused on the conserved patterns of the G protein-coupled receptors, an important superfamily of receptors. Olfactory receptors represent more than 2% of our genome and constitute the largest family of G protein-coupled receptors, a key class of drug targets. As no crystallographic structures are available, mechanistic studies rely on the use of molecular dynamic modelling combined with site-directed mutagenesis data. In this paper, we hypothesized that human-mouse orthologs coding for G protein-coupled receptors maintain, at speciation events, shared compositional structures independent, to some extent, of their percent identity as reveals a method based in the categorization of nucleotide triplets by their gross composition. The data support the consistency of the hypothesis, showing in ortholog G protein-coupled receptors the presence of emergent shared compositional structures preserved at speciation events. -

WO 2019/068007 Al Figure 2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/068007 Al 04 April 2019 (04.04.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors; and C12N 15/10 (2006.01) C07K 16/28 (2006.01) (71) Applicants: GROSS, Gideon [EVIL]; IE-1-5 Address C12N 5/10 (2006.0 1) C12Q 1/6809 (20 18.0 1) M.P. Korazim, 1292200 Moshav Almagor (IL). GIBSON, C07K 14/705 (2006.01) A61P 35/00 (2006.01) Will [US/US]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., C07K 14/725 (2006.01) P.O. Box 4044, 7403635 Ness Ziona (TL). DAHARY, Dvir [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (21) International Application Number: Box 4044, 7403635 Ness Ziona (IL). BEIMAN, Merav PCT/US2018/053583 [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (22) International Filing Date: Box 4044, 7403635 Ness Ziona (E.). 28 September 2018 (28.09.2018) (74) Agent: MACDOUGALL, Christina, A. et al; Morgan, (25) Filing Language: English Lewis & Bockius LLP, One Market, Spear Tower, SanFran- cisco, CA 94105 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/564,454 28 September 2017 (28.09.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/649,429 28 March 2018 (28.03.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (71) Applicant: IMMP ACT-BIO LTD. -

Common Homozygosity for Predicted Loss-Of-Function Variants Reveals Both Redundant and Advantageous Effects of Dispensable Human Genes
Common homozygosity for predicted loss-of-function variants reveals both redundant and advantageous effects of dispensable human genes. Antonio Rausell, Yufei Luo, Marie Lopez, Yoann Seeleuthner, Franck Rapaport, Antoine Favier, Peter Stenson, David Cooper, Etienne Patin, Jean-Laurent Casanova, et al. To cite this version: Antonio Rausell, Yufei Luo, Marie Lopez, Yoann Seeleuthner, Franck Rapaport, et al.. Common homozygosity for predicted loss-of-function variants reveals both redundant and advantageous effects of dispensable human genes.. Proceedings of the National Academy of Sciences of the United States of America , National Academy of Sciences, 2020, 117 (24), pp.13626-13636. 10.1073/pnas.1917993117. hal-03020429 HAL Id: hal-03020429 https://hal.archives-ouvertes.fr/hal-03020429 Submitted on 18 Dec 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ArticleExpress Dear Author Please use this PDF proof to check the layout of your article. If you would like any changes to be made to the layout, you can leave instructions in the online proofing interface. Making your changes directly in the online proofing interface is the quickest, easiest way to correct and submit your proof. Please note that changes made to the article in the online proofing interface will be added to the article before publication, but are not reflected in this PDF proof. -
Onderstaande Coverage Is Berekend Over 860 Exomen, Welke Geprept Zijn Met De Agilent Sureselect XT Exome V7 Kit
Onderstaande coverage is berekend over 860 exomen, welke geprept zijn met de Agilent SureSelect XT exome v7 kit. Het sequencen is uitgevoerd op een Illumina NextSeq500 of NovaSeq6000 met een gemiddelde coverage van 100X , dekking 20x >93%. Gemiddelde Gen Coverage 20x A1BG 128 99.8 A1CF 132 99.32 A2ML1 155 99.97 A2M 148 99.72 A3GALT2 119 100 A4GALT 157 100 A4GNT 178 100 AAAS 123 100 AACS 143 97.96 AADACL2 186 99.9 AADACL3 161 100 AADACL4 170 100 AADAC 160 99.98 AADAT 138 97.39 AAED1 110 84.89 AAGAB 164 99.29 AAK1 122 99.39 AAMDC 135 90 AAMP 95 98.4 AANAT 86 99.85 AAR2 120 98.41 AARD 75 99.06 AARS2 123 99.89 AARSD1 115 94.24 AARS 134 100 AASDHPPT 157 99.75 AASDH 135 99.29 AASS 143 99.67 AATF 156 99.95 AATK 79 92.71 ABAT 136 99.16 ABCA1 149 100 ABCA2 123 96.52 ABCA3 132 99.27 ABCA4 137 96.03 ABCA5 101 94.3 ABCA6 131 98.41 ABCA7 102 99.12 ABCA8 146 99.26 ABCA9 140 99.34 ABCA10 126 94.11 ABCA12 159 99.8 ABCA13 159 97.2 ABCB1 160 99.58 ABCB4 142 99.43 ABCB5 151 99.76 ABCB6 149 99.98 ABCB7 128 99.81 ABCB8 100 93.36 ABCB9 124 97.14 ABCB10 136 90.97 ABCB11 158 99.22 ABCC1 141 96.65 ABCC2 150 99.71 ABCC3 108 96.16 ABCC4 134 95.97 ABCC5 130 96.14 ABCC6 96 92.85 ABCC8 136 99.83 ABCC9 141 99.75 ABCC10 90 99.35 ABCC11 117 99.93 ABCC12 153 99.93 ABCD1 74 83.58 ABCD2 143 99.92 ABCD3 122 92.63 ABCD4 122 99.84 ABCE1 141 98.18 ABCF1 90 99.42 ABCF2 128 99.93 ABCF3 159 100 ABCG1 135 99.71 ABCG2 143 99.77 ABCG4 122 99.91 De Coverage komt vanuit LAB-F0680_Exoom Coverage_v3. -

Gnomad Lof Supplement
1 gnomAD supplement gnomAD supplement 1 Data processing 4 Alignment and read processing 4 Variant Calling 4 Coverage information 5 Data processing 5 Sample QC 7 Hard filters 7 Supplementary Table 1 | Sample counts before and after hard and release filters 8 Supplementary Table 2 | Counts by data type and hard filter 9 Platform imputation for exomes 9 Supplementary Table 3 | Exome platform assignments 10 Supplementary Table 4 | Confusion matrix for exome samples with Known platform labels 11 Relatedness filters 11 Supplementary Table 5 | Pair counts by degree of relatedness 12 Supplementary Table 6 | Sample counts by relatedness status 13 Population and subpopulation inference 13 Supplementary Figure 1 | Continental ancestry principal components. 14 Supplementary Table 7 | Population and subpopulation counts 16 Population- and platform-specific filters 16 Supplementary Table 8 | Summary of outliers per population and platform grouping 17 Finalizing samples in the gnomAD v2.1 release 18 Supplementary Table 9 | Sample counts by filtering stage 18 Supplementary Table 10 | Sample counts for genomes and exomes in gnomAD subsets 19 Variant QC 20 Hard filters 20 Random Forest model 20 Features 21 Supplementary Table 11 | Features used in final random forest model 21 Training 22 Supplementary Table 12 | Random forest training examples 22 Evaluation and threshold selection 22 Final variant counts 24 Supplementary Table 13 | Variant counts by filtering status 25 Comparison of whole-exome and whole-genome coverage in coding regions 25 Variant annotation 30 Frequency and context annotation 30 2 Functional annotation 31 Supplementary Table 14 | Variants observed by category in 125,748 exomes 32 Supplementary Figure 5 | Percent observed by methylation. -

Genetic Variants Affecting Equivalent Protein Family Positions Reflect
www.nature.com/scientificreports OPEN Genetic variants afecting equivalent protein family positions refect human diversity Received: 13 January 2017 Francesco Raimondi1,2, Matthew J. Betts1,2, Qianhao Lu1,2, Asuka Inoue3,4, J. Silvio Gutkind5 & Accepted: 13 September 2017 Robert B. Russell 1,2 Published: xx xx xxxx Members of diverse protein families often perform overlapping or redundant functions meaning that diferent variations within them could refect diferences between individual organisms. We investigated likely functional positions within aligned protein families that contained a signifcant enrichment of nonsynonymous variants in genomes of healthy individuals. We identifed more than a thousand enriched positions across hundreds of family alignments with roles indicative of mammalian individuality, including sensory perception and the immune system. The most signifcant position is the Arginine from the Olfactory receptor “DRY” motif, which has more variants in healthy individuals than all other positions in the proteome. Odorant binding data suggests that these variants lead to receptor inactivity, and they are mostly mutually exclusive with other loss-of-function (stop/frameshift) variants. Some DRY Arginine variants correlate with smell preferences in sub-populations and all 2,504 humans studied contain a unique spectrum of active and inactive receptors. The many other variant enriched positions, across hundreds of other families might also provide insights into individual diferences. Genomes from healthy individuals1 illuminate both diseases2 and attributes of individuals or human popula- tions3,4. Perhaps the most common current use of these genomes is to provide background mutation rates to assess candidate disease mutations or to identify likely deleterious genetic alterations5,6. For example, ExAC6 is frequently used by human geneticists to determine variant frequency during searches for disease causing mutations. -

Amino Acid Sequences Directed Against Cxcr4 And
(19) TZZ ¥¥_T (11) EP 2 285 833 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: C07K 16/28 (2006.01) A61K 39/395 (2006.01) 17.12.2014 Bulletin 2014/51 A61P 31/18 (2006.01) A61P 35/00 (2006.01) (21) Application number: 09745851.7 (86) International application number: PCT/EP2009/056026 (22) Date of filing: 18.05.2009 (87) International publication number: WO 2009/138519 (19.11.2009 Gazette 2009/47) (54) AMINO ACID SEQUENCES DIRECTED AGAINST CXCR4 AND OTHER GPCRs AND COMPOUNDS COMPRISING THE SAME GEGEN CXCR4 UND ANDERE GPCR GERICHTETE AMINOSÄURESEQUENZEN SOWIE VERBINDUNGEN DAMIT SÉQUENCES D’ACIDES AMINÉS DIRIGÉES CONTRE CXCR4 ET AUTRES GPCR ET COMPOSÉS RENFERMANT CES DERNIÈRES (84) Designated Contracting States: (74) Representative: Hoffmann Eitle AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Patent- und Rechtsanwälte PartmbB HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL Arabellastraße 30 PT RO SE SI SK TR 81925 München (DE) (30) Priority: 16.05.2008 US 53847 P (56) References cited: 02.10.2008 US 102142 P EP-A- 1 316 801 WO-A-99/50461 WO-A-03/050531 WO-A-03/066830 (43) Date of publication of application: WO-A-2006/089141 WO-A-2007/051063 23.02.2011 Bulletin 2011/08 • VADAY GAYLE G ET AL: "CXCR4 and CXCL12 (73) Proprietor: Ablynx N.V. (SDF-1) in prostate cancer: inhibitory effects of 9052 Ghent-Zwijnaarde (BE) human single chain Fv antibodies" CLINICAL CANCER RESEARCH, THE AMERICAN (72) Inventors: ASSOCIATION FOR CANCER RESEARCH, US, • BLANCHETOT, Christophe vol.10, no. -

Multivariate GWAS of Psychiatric Disorders and Their Cardinal Symptoms Reveal Two Dimensions of Cross-Cutting Genetic Liabilities
bioRxiv preprint doi: https://doi.org/10.1101/603134; this version posted August 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Multivariate GWAS of psychiatric disorders and their cardinal symptoms reveal two dimensions of cross-cutting genetic liabilities Travis T. Mallard1, Richard K. Linnér2,3, Andrew D. Grotzinger1, Sandra Sanchez-Roige4, Jakob Seidlitz5,6, Aysu Okbay2, Ronald de Vlaming2, S. Fleur W. Meddens7, Bipolar Disorder Working Group of the Psychiatric Genomics Consortium^, Abraham A. Palmer4,8, Lea K. Davis9,10, Elliot M. Tucker-Drob1,11, Kenneth S. Kendler12,13, Matthew C. Keller14,15, Philipp D. Koellinger2*, & K. Paige Harden1,11* ^Full list of consortium authors is reported in Supplementary Note. *Work was jointly supervised by these authors. Affiliations: 1. Department of Psychology, University of Texas at Austin, Austin, TX, USA 2. Department of Economics, School of Business and Economics, Vrije Universiteit Amsterdam, Amsterdam, The Netherlands 3. Autism and Developmental Medicine Institute, Geisinger, Lewisburg, PA, USA 4. Department of Psychiatry, University of California San Diego, La Jolla, CA, USA 5. Department of Child and Adolescent Psychiatry and Behavioral Science, Children's Hospital of Philadelphia, Philadelphia, PA, USA 6. Department of Psychiatry, University of Pennsylvania, Philadelphia, PA, USA 7. Erasmus University Rotterdam Institute for Behavior and Biology, Erasmus School of Economics, Erasmus University Rotterdam, Rotterdam, The Netherlands 8. Institute for Genomic Medicine, University of California San Diego, La Jolla, CA, USA 9. -

From Musk to Body Odor: Decoding Olfaction Through Genetic Variation
bioRxiv preprint doi: https://doi.org/10.1101/2021.04.27.441177; this version posted April 28, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. From musk to body odor: decoding olfaction through genetic variation Bingjie Li1,2,*, Marissa L. Kamarck3,4,*, Qianqian Peng1,*, Fei-Ling Lim5, Andreas Keller6, Monique A.M. Smeets7, Joel D. Mainland3,4,a, and Sijia Wang1,8,a 1CAS Key Laboratory of Computational Biology, Shanghai Institute of Nutrition and Health, University of Chinese Academy of Sciences, Chinese Academy of Sciences, China; 2Department of Skin and Cosmetics Research, Shanghai Skin Disease Hospital, Tongji University School of Medicine, Shanghai, China; 3Monell Chemical Senses Center, Philadelphia, PA 19104, USA; 4Department of Neuroscience, University of Pennsylvania, Philadelphia, PA 19104, USA; 5Unilever Research & Development, Colworth, UK; 6Laboratory of Neurogenetics and Behavior, The Rockefeller University, New York, NY 10065 USA; 7Unilever Research & Development, Rotterdam, The Netherlands; 8Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming 650223, China This manuscript was compiled on April 23, 2021 The olfactory system combines input from multiple receptor types assay(5–12). to represent odor information, but there are few explicit examples Here, we utilize the same strategy of correlating perceptual relating olfactory receptor (OR) activity patterns to odor perception. and genetic variation, but with three improvements: 1. Using To uncover these relationships, we performed genome-wide scans a larger population to increase power, 2. -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -

11 22 33 44 55 Aa
bip_pgc3.6.chr3 20 1 0.8 0.6 0.4 0.2 0.1 1 . rs9834970 : Bipolar_disorder(22182935)(1E−12) snp / p / or / maf / info / directions Schizophrenia_o...(24280982)(1E−10)1 a a . rs9834970 / 6.63e−19 / 1.09 / 0.48 / 0.992 / 13−44−0 Bipolar_disorder(28072414)(2E−10) ● Bipolar_disorder(27329760)(5E−10)●●● ●● ●● Bipolar_disorder(28115744)(2E−9) 15 40 2 . rs3732386 : ●● Schizophrenia(28991256)(3E−11) ●● ● 3 . rs6550435 : Bipolar_disorder(24618891)(2E−8)●● ●● ● ● ● ● ● ● ●● ●● 4 . rs75968099 : Schizophrenia(25056061)(1E−13) ● ●● ●● Schizophrenia(26198764)(2E−12) ●● ● ● ● ●● 10 Autism_spectrum...(28540026)(1E−11)●● ● ● ● ●● ●●● ●● ●● ●● ●● ● ● ● ● ●● ● ● ● 5 . rs6789885 : Platelet_distri...(27863252)(2E−10)● ●● ●●●● 2 ● ●● 32 ● ● ●● ●● 3 ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● 4● ●● ● ● p = 5.0e−08 ●● ● ●● ● ● ● ● ●● 20 ●● ● ● ● ●● ●● Observed (−logP) ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ●● ● ● ●● ●● ● ●● ● ● ● ●●●● ●● ● ● ●● ● ● ● ●● ● ● ● 5 ● ●● ● ● ●● ●● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ●● ● ● ● ● ●● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●●●●● ● ● ● ● ●● ● ● 5● ● ● ● ● ● ● 5 ● ● ● ● ● ●● ● ● ●● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ●● ● ● ●●●● ●● ● ● ● ●● ●●● ● ● ● ●●● ● ●● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ●● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ●●● ● ● ● ●● ● ●● ● ● ● ●● ● ●● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ● ● ● ● ●● ● ● ● ● ● ●● ● ● ● ● ● ● ● ●● ● ● ● ● ● ● ● ● ● ● ●