Giving Molecules an Identity. on the Interplay Between Qsars and Partial Order Ranking
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

No. Name Major Application 1 ALDICARB Pesticide/Insecticide
Index No. Name Major application 1 ALDICARB Pesticide/Insecticide, Acaricide, Nematicide 2 AMITRAZ Pesticide/Insecticide, Acaricide 3 BIFENTHRIN Pesticide/Insecticide, Acaricide 4 BIORESMETHRIN Pesticide/Insecticide 5 BROMIDE Pesticide/Insecticide 6 CARBARYL Pesticide/Insecticide/Plant growth regulator 7 CHLORDANE Pesticide/Insecticide 8 CHLORMEQUAT Pesticide/Plant growth regulator 9 CHLORPYRIFOS Pesticide/Insecticide 10 CHLORPYRIFOS-METHYL Pesticide/Insecticide, Acaricide 11 CLETHODIM Pesticide/Herbicide 12 CYHALOTHRIN Pesticide&Veterinary Drugs/Insecticide 13 CYPERMETHRIN Pesticide&Veterinary Drugs/Insecticide 14 DELTAMETHRIN Pesticide&Veterinary Drugs/Insecticide 15 DIAZINON Pesticide&Veterinary Drugs/Insecticide, Acaricide 16 DICHLORVOS、NALED Pesticide&Veterinary Drugs/Insecticide 17 DICOFOL Pesticide/Insecticide, Acaricide 18 DIMETHIPIN Pesticide/Herbicide/Plant growth regulator 19 DIMETHOATE Pesticide/Insecticide, Acaricide 20 DIPHENYLAMINE Pesticide/Bactericide 21 DIQUAT Pesticide/Herbicide 22 ENDOSULFAN Pesticide/Insecticide, Acaricide 23 ETHEPHON Pesticide/Plant growth regulator 24 FENARIMOL Pesticide/Bactericide 25 FENBUTATIN OXIDE Pesticide/Insecticide, Acaricide 26 FENITROTHION Pesticide&Veterinary Drugs/Insecticide 27 FENPROPATHRIN Pesticide/Insecticide, Acaricide 28 FENTHION Pesticide/Insecticide 29 FENVALERATE Pesticide&Veterinary Drugs/Insecticide, Acaricide 30 FLUSILAZOLE Pesticide/Bactericide 31 GLUFOSINATE Pesticide/Herbicide 32 GLYPHOSATE(SULFOSATE) Pesticide/Herbicide 33 HEPTACHLOR Pesticide/Insecticide 34 HYDROGEN -

Organophosphate Poisoning : a Review
120 Sinha and Sharma Med J Indones Organophosphate poisoning : A review Parmod K. Sinha, Ashok Sharma Abstrak Pestisida organofosfat digunakan secara luas di seluruh dunia. Keracunan oleh bahan ini merupakan masalah kesehatan masyarakat, terutama di negara berkembang. Zat neurotoksik organofosfat merupakan bahan yang dianggap mengancam dalam bidang militer dan terorisme. Mekanisme toksisitas bahan ini adalah dengan cara menghambat asetilkolinesterase yang mengakibatkan menumpuknya neurotransmitor asetilkolin dan terjadi rangsangan terus-menerus pada reseptor asetilkolin pada sistem saraf sentral maupun perifer. Selain krisis kolinergik, organofosfat dapat menimbulkan berbagai sindrom neurologis, baik akut maupun kronik. Sedangkan gejala peralihan ( intermediate) terjadi 1-4 hari setelah krisis kolinergik teratasi. Pengobatan standar terdiri dari reaktivasi asetilkolinesterase dengan antidot golongan oksim (prolidoksim, oksidoksime, HI-6 dan HLo7), dan pengendalian efek biokimia asetilkolin dengan menggunakan atropin. Golongan oksim yang baru HI-6 dan Hlo7 merupakan reaktivator asetilkolinesterase yang lebih cocok dan efektif untuk keracunan akut dan berat dibandingkan dengan prolidoksim dan obidoksim. Penderita yang mendapat pengobatan segera, biasanya dapat sembuh dari toksisitas akut, namun gejala neurologis ikutan dapat saja terjadi. (Med J Indones 2003; 12: 120-6) Abstract Organophosphate pesticides are used extensively worldwide, and poisoning by these agents, particularly in developing nations is a public health problem. Organophosphorous -

Chemical Name Federal P Code CAS Registry Number Acutely
Acutely / Extremely Hazardous Waste List Federal P CAS Registry Acutely / Extremely Chemical Name Code Number Hazardous 4,7-Methano-1H-indene, 1,4,5,6,7,8,8-heptachloro-3a,4,7,7a-tetrahydro- P059 76-44-8 Acutely Hazardous 6,9-Methano-2,4,3-benzodioxathiepin, 6,7,8,9,10,10- hexachloro-1,5,5a,6,9,9a-hexahydro-, 3-oxide P050 115-29-7 Acutely Hazardous Methanimidamide, N,N-dimethyl-N'-[2-methyl-4-[[(methylamino)carbonyl]oxy]phenyl]- P197 17702-57-7 Acutely Hazardous 1-(o-Chlorophenyl)thiourea P026 5344-82-1 Acutely Hazardous 1-(o-Chlorophenyl)thiourea 5344-82-1 Extremely Hazardous 1,1,1-Trichloro-2, -bis(p-methoxyphenyl)ethane Extremely Hazardous 1,1a,2,2,3,3a,4,5,5,5a,5b,6-Dodecachlorooctahydro-1,3,4-metheno-1H-cyclobuta (cd) pentalene, Dechlorane Extremely Hazardous 1,1a,3,3a,4,5,5,5a,5b,6-Decachloro--octahydro-1,2,4-metheno-2H-cyclobuta (cd) pentalen-2- one, chlorecone Extremely Hazardous 1,1-Dimethylhydrazine 57-14-7 Extremely Hazardous 1,2,3,4,10,10-Hexachloro-6,7-epoxy-1,4,4,4a,5,6,7,8,8a-octahydro-1,4-endo-endo-5,8- dimethanonaph-thalene Extremely Hazardous 1,2,3-Propanetriol, trinitrate P081 55-63-0 Acutely Hazardous 1,2,3-Propanetriol, trinitrate 55-63-0 Extremely Hazardous 1,2,4,5,6,7,8,8-Octachloro-4,7-methano-3a,4,7,7a-tetra- hydro- indane Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]- 51-43-4 Extremely Hazardous 1,2-Benzenediol, 4-[1-hydroxy-2-(methylamino)ethyl]-, P042 51-43-4 Acutely Hazardous 1,2-Dibromo-3-chloropropane 96-12-8 Extremely Hazardous 1,2-Propylenimine P067 75-55-8 Acutely Hazardous 1,2-Propylenimine 75-55-8 Extremely Hazardous 1,3,4,5,6,7,8,8-Octachloro-1,3,3a,4,7,7a-hexahydro-4,7-methanoisobenzofuran Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime 26419-73-8 Extremely Hazardous 1,3-Dithiolane-2-carboxaldehyde, 2,4-dimethyl-, O- [(methylamino)-carbonyl]oxime. -

Malathion Human Health and Ecological Risk Assessment Final Report
SERA TR-052-02-02c Malathion Human Health and Ecological Risk Assessment Final Report Submitted to: Paul Mistretta, COR USDA/Forest Service, Southern Region 1720 Peachtree RD, NW Atlanta, Georgia 30309 USDA Forest Service Contract: AG-3187-C-06-0010 USDA Forest Order Number: AG-43ZP-D-06-0012 SERA Internal Task No. 52-02 Submitted by: Patrick R. Durkin Syracuse Environmental Research Associates, Inc. 5100 Highbridge St., 42C Fayetteville, New York 13066-0950 Fax: (315) 637-0445 E-Mail: [email protected] Home Page: www.sera-inc.com May 12, 2008 Table of Contents Table of Contents............................................................................................................................ ii List of Figures................................................................................................................................. v List of Tables ................................................................................................................................. vi List of Appendices ......................................................................................................................... vi List of Attachments........................................................................................................................ vi ACRONYMS, ABBREVIATIONS, AND SYMBOLS ............................................................... vii COMMON UNIT CONVERSIONS AND ABBREVIATIONS.................................................... x CONVERSION OF SCIENTIFIC NOTATION .......................................................................... -

Qsar Analysis of the Chemical Hydrolysis of Organophosphorus Pesticides in Natural Waters
QSAR ANALYSIS OF THE CHEMICAL HYDROLYSIS OF ORGANOPHOSPHORUS PESTICIDES IN NATURAL WATERS. by Kenneth K. Tanji Principal Investigator and Jonathan 1. Sullivan Graduate Research Assistant Department of Land, Air and Water Resources University of California, Davis Technical Completion Report Project Number W-843 August, 1995 University of California Water Resource Center The research leading to this report was supported by the University of California Water Resource Center as part of Water Resource Center Project W-843. Table of Contents Page Abstract 2 Problem and Research Objectives 3 Introduction 5 Theoretical Background 6 QSAR Methodology 7 Molecular Connectivity Theory 8 Organophosphorus Pesticides 12 Experimental Determination of Rates 15 Results and Discussion 17 Principal Findings and Significance 19 References 34 List of Tables Page Table 1. Statistical relationship between OP pesticides and first-order MC/'s. 30 Table 2. Inherent conditions of waters used in experimental work. 16 Table 3. Estimated half-lives for organophosphorus esters derived from model. 31 Table 4. Half-lives and first-order MCI' sfor model calibration data set. 31 Table 5. Experimental kinetic data for validation set compounds, Sacramento. 33 List of Figures Page Figure 1. Essential Features OfQSAR Modeling Methodology. 21 Figure 2. Regression plot for In hydrolysis rate vs. 1st order MCl' s. 22 Figure 3. a 3-D molecular model, a line-segment model and a graphical model. 23 Figure 4. Molecular connectivity index suborders. 24 Figure 5. Chlorpyrifos and its fourteen fourth order path/cluster fragments. 25 Figure 6. Abridged MClndex output. 26 Figure 7. Parent acids of most common organophosphorus pesticides. 12 Figure 8. -

Environmental Health Criteria 63 ORGANOPHOSPHORUS
Environmental Health Criteria 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION Please note that the layout and pagination of this web version are not identical with the printed version. Organophophorus insecticides: a general introduction (EHC 63, 1986) INTERNATIONAL PROGRAMME ON CHEMICAL SAFETY ENVIRONMENTAL HEALTH CRITERIA 63 ORGANOPHOSPHORUS INSECTICIDES: A GENERAL INTRODUCTION This report contains the collective views of an international group of experts and does not necessarily represent the decisions or the stated policy of the United Nations Environment Programme, the International Labour Organisation, or the World Health Organization. Published under the joint sponsorship of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization World Health Orgnization Geneva, 1986 The International Programme on Chemical Safety (IPCS) is a joint venture of the United Nations Environment Programme, the International Labour Organisation, and the World Health Organization. The main objective of the IPCS is to carry out and disseminate evaluations of the effects of chemicals on human health and the quality of the environment. Supporting activities include the development of epidemiological, experimental laboratory, and risk-assessment methods that could produce internationally comparable results, and the development of manpower in the field of toxicology. Other activities carried out by the IPCS include the development of know-how for coping with chemical accidents, coordination -
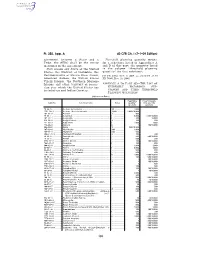
40 CFR Ch. I (7–1–09 Edition) Pt. 355, App. A
Pt. 355, App. A 40 CFR Ch. I (7–1–09 Edition) agreement between a State and a Threshold planning quantity means, Tribe, the SERC shall be the entity for a substance listed in Appendices A identified in the agreement. and B of this part, the quantity listed State means any State of the United in the column ‘‘threshold planning States, the District of Columbia, the quantity’’ for that substance. Commonwealth of Puerto Rico, Guam, [73 FR 65462, Nov. 3, 2008, as amended at 73 American Samoa, the United States FR 76960, Dec. 18, 2008] Virgin Islands, the Northern Mariana Islands, any other territory or posses- APPENDIX A TO PART 355—THE LIST OF sion over which the United States has EXTREMELY HAZARDOUS SUB- jurisdiction and Indian Country. STANCES AND THEIR THRESHOLD PLANNING QUANTITIES [Alphabetical Order] Reportable Threshold plan- CAS No. Chemical name Notes quantity * ning quantity (pounds) (pounds) 75–86–5 ................ Acetone Cyanohydrin ................................................. 10 ................ 1,000 1752–30–3 ............ Acetone Thiosemicarbazide ....................................... 1,000 ........... 1,000/10,000 107–02–8 .............. Acrolein ....................................................................... 1 .................. 500 79–06–1 ................ Acrylamide .................................................................. f ................... 5,000 1,000/10,000 107–13–1 .............. Acrylonitrile ................................................................. f ................... 100 10,000 -

Appendix Common, Trade, and Chemical Names of Pesticides Mentioned in the Present Volume
Appendix Common, trade, and chemical names of pesticides mentioned in the present volume Commonname Tradename Chemical name aldrin Octalene 1,2,3,4,10,1 O-hexachloro-1 ,4,4a,S,8,8a hexahydro-1,4-endo-exo-S,8-dimethano naphthalene amidithion Thiocron O,O-dimethyl S-(2-methoxyethyl carbamoylmethyl) phosphorodithioate azinphos methyl Guthion, Gusathion O,O-dimcthyl S-( 4-oxo-l ,2,3-benzotri azin-3-( 4H)-ylmethyl) phosphorodithioate captan Orthocid N -(trichloromethylthio) cyclohex-4-ene- 1,2-dicarboximide carbaryl Sevin 1-naphthyl-mcthylcarbamate carbophenothion Trithion O,O-diethyl S-[(p-chlorophenylthio) methyl] phosphorodithioate chlorfenvinphos Birlane (Shell), 2-chloro-1-(2,4-dichlorophenyl) vinyl Sapecron (e/BA) diethyl phosphate chlorphenamidine Galecron N'-( 4-chloro-o-tolyl)-N,N-dimethyl formamidine chlorthion Chlorthion O,O-dimethyIO-(3-chloro-4-nitro phenyl)-phosphorothioate coumaphos Asuntol, Co-Ral O,O-diethyl O-(3-chloro-4-methyl- 2-oxo-2H-1-benzopyran-7-yl) phosphorothioate DDT Gesarol 1,1,1-trichloro-2,2-bis(p-chlorophenyl) ethane demeton Systox O,O-diethyl O-(and S)-2-(ethylthio)ethyl phosphorothioates DEF S,S,S-tributyltrithiophosphate demeton methyl Metasystox O,O-dimethyl O-(and S)-2-(ethylthio) ethyl phosphorothioates diazinon Diazinon, Basudin O,O-diethyIO-(2-isopropyl-4-methyl- 6-pyrimidyl) phosphorothioate dichlorvos Vapona (Shell), O,O-dimethyl-2,2-dichlorovinyl Nuvan (e/BA) phosphate dicrotophos Bidrin (Shell), O,O-dimethyl O-(2-dimethyl-carbamyl-1- Carbicron (e/BA) methyl) vinyl phosphate dieldrin Octalox -

Phosphamidon
Phosphamidon sc-250712 Material Safety Data Sheet Hazard Alert Code EXTREME HIGH MODERATE LOW Key: Section 1 - CHEMICAL PRODUCT AND COMPANY IDENTIFICATION PRODUCT NAME Phosphamidon STATEMENT OF HAZARDOUS NATURE CONSIDERED A HAZARDOUS SUBSTANCE ACCORDING TO OSHA 29 CFR 1910.1200. NFPA FLAMMABILITY1 HEALTH4 HAZARD INSTABILITY0 SUPPLIER Santa Cruz Biotechnology, Inc. 2145 Delaware Avenue Santa Cruz, California 95060 800.457.3801 or 831.457.3800 EMERGENCY ChemWatch Within the US & Canada: 877-715-9305 Outside the US & Canada: +800 2436 2255 (1-800-CHEMCALL) or call +613 9573 3112 SYNONYMS C10-H19-Cl-N-O5-P, CH3CH2)2N(O)CCCl=C(CH3)OP(=)(OCH3)2, "phosphoric acid, ", "2-chloro-2-(diethylamino)-1-methyl-3-oxo- 1-propenyl dimethyl ester", "phosphoric acid, dimethyl ester, ", "ester with 2-chloro-N, N-diethyl-3-hydroxycrotonamide", "2-chloro- 2-diethylcarbamoyl-1-methylvinyl dimethylphosphate", "1-chloro-diethylcarbamoyl-1-propen-2-yl dimethyl phosphate", "2-chloro- 2-dimethylcarbamoyl-1-methylvinyl dimethyl phosphate", Apamidon, C570, "CIBA 570", Dimecron, "Dimecron 100", Dixon, Famfos, Fosfamidone, Merkon, Phosphamidone, "OMS 1325", "OR 1191" Section 2 - HAZARDS IDENTIFICATION CHEMWATCH HAZARD RATINGS Min Max Flammability: 1 Toxicity: 4 Body Contact: 3 Min/Nil=0 Low=1 Reactivity: 1 Moderate=2 High=3 Chronic: 2 Extreme=4 CANADIAN WHMIS SYMBOLS 1 of 12 EMERGENCY OVERVIEW RISK Toxic in contact with skin. Possible risk of irreversible effects. Very toxic by inhalation and if swallowed. Very toxic to aquatic organisms, may cause long-term adverse effects in the aquatic environment. Cumulative effects may result following exposure*. May produce discomfort of the eyes*. Limited evidence of a carcinogenic effect*. -

1 of 3 GC+LC-USA
Updated: 07/18/2016 1 of 3 GC+LC-USA Limit of Quantitation (LOQ): 0.010 mg/kg (ppm) Sample Types: Low Fat Content Samples Minimum Sample Size: 100 grams (~1/4 pound). Certain products require more for better sample representation. Instrument: GC-MS/MS and LC-MS/MS Turnaround: 24-48 hours Accreditation: Part of AGQ USA's ISO/IEC 17025 Accreditation Scope 4,4'-Dichlorobenzophenone Bupirimate Cyantraniliprole Diflufenican Abamectin Buprofezin Cyazofamid Dimethoate Acephate Butachlor Cycloate Dimethoate (Sum) Acequinocyl Butocarboxim Cycloxydim Dimethomorph Acetamiprid Butralin Cyflufenamid Diniconazole Acetochlor Cadusafos Cyfluthrin Dinocap Acrinathrin Captafol Cymoxanil Dinotefuran Alachlor Captan Cyproconazole Diphenylamine Aldicarb Captan (Sum) Cyprodinil Disulfoton Aldicarb (Sum) Carbaryl Cyromazine Disulfoton (Sum) Aldicarb-sulfone Carbofuran DDD-o,p Disulfoton-sulfone Aldicarb-sulfoxide Carbofuran-3-hydroxy DDD-p,p +DDT-o,p Disulfoton-sulfoxide Aldrin Carbophenothion DDE-o,p Ditalimfos Ametryn Carbosulfan DDE-p,p Diuron Amitraz Carboxine DDT (Sum) Dodemorph Atrazine Carfentrazone-ethyl DDT-p,p Dodine Azadirachtin Chinomethionat DEET Emamectin Benzoate Azamethiphos Chlorantraniliprole Deltamethrin Endosulfan (A+B+Sulf) Azinphos-ethyl Chlordane Demeton Endosulfan Alfa Azinphos-methyl Chlordane Trans Demeton-S-methyl-sulfone Endosulfan Beta Azoxystrobin Chlorfenapyr Desmedipham Endosulfan Sulfate Benalaxyl Chlorfenson Diafenturion Endrin Ben-Carb-TPM (Sum) Chlorfenvinphos Dialifos EPN Bendiocarb Chlorfluazuron Diazinon Epoxiconazole -

NMP-Free Formulations of Neonicotinoids
(19) & (11) EP 2 266 400 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 29.12.2010 Bulletin 2010/52 A01N 43/40 (2006.01) A01N 43/86 (2006.01) A01N 47/40 (2006.01) A01N 51/00 (2006.01) (2006.01) (2006.01) (21) Application number: 09305544.0 A01P 7/00 A01N 25/02 (22) Date of filing: 15.06.2009 (84) Designated Contracting States: (72) Inventors: AT BE BG CH CY CZ DE DK EE ES FI FR GB GR • Gasse, Jean-Jacques HR HU IE IS IT LI LT LU LV MC MK MT NL NO PL 27600 Saint-Aubin-Sur-Gaillon (FR) PT RO SE SI SK TR • Duchamp, Guillaume Designated Extension States: 92230 Gennevilliers (FR) AL BA RS • Cantero, Maria 92230 Gennevilliers (FR) (71) Applicant: NUFARM 92233 Gennevelliers (FR) (74) Representative: Cabinet Plasseraud 52, rue de la Victoire 75440 Paris Cedex 09 (FR) (54) NMP-free formulations of neonicotinoids (57) The invention relates to NMP-free liquid formulation comprising at least one nicotinoid and at least one aprotic polar component selected from the group comprising the compounds of formula I, II or III below, and mixtures thereof, wherein R1 and R2 independently represent H or an alkyl group having less than 5 carbons, preferably a methyl group, and n represents an integer ranging from 0 to 5, and to their applications. EP 2 266 400 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 266 400 A1 Description Technical Field of the invention 5 [0001] The invention relates to novel liquid formulations of neonicotinoids and to their use for treating plants, for protecting plants from pests and/or for controlling pests infestation. -

China Releases New Maximum Residue Limits for Pesticides In
GB 2763-2016 THIS REPORT CONTAINS ASSESSMENTS OF COMMODITY AND TRADE ISSUES MADE BY USDA STAFF AND NOT NECESSARILY STATEMENTS OF OFFICIAL U.S. GOVERNMENT POLICY Voluntary - Public Date: 3/31/2017 GAIN Report Number: CH17016 China - Peoples Republic of Post: Beijing China Releases New Maximum Residue Limits for Pesticides in Food Report Categories: FAIRS Subject Report Approved By: Lisa Anderson Prepared By: FAS Staff Report Highlights: On December 18, 2016, the Chinese National Health and Family Planning Commission, Ministry of Agriculture, China Food and Drug Administration released the National Food Safety Standard - Maximum Residue Limits for Pesticides in Foods (GB 2763-2016). The standard will replace the current MRL Standard (GB 2763-2014) and will be implemented on June 18, 2017. This report provides an unofficial translation of the standard. Editors’ Note: The asterisk appearing in the MRL column means that the limit is a temporary MRL. A temporary MRL is usually set under the following four conditions: 1. The dietary risk assessment data is incomplete; 2. The Acceptable Daily Intake (ADI) is temporary (ADI is used as the basis for MRL setting); 3. There is no surveillance or analysis method for the MRL that complies with the standard requirements; 4. In emergency situations, the pesticide is approved to be used on un-registered crops. I GB 2763-2016 General Information: BEGIN TRANSLATION ICS 65.100 G 25 GB National Standard of the People’s Republic of China GB 2763—2016 Replacing GB 2763 - 2014 National food safety standard Maximum Residue Limits for Pesticides in Food General Information: National Health and Family Planning Commission Issued by: Ministry of Agriculture China Food and Drug Administration Issued on: 2016-12-18 Implementation:2017-06-18 II GB 2763-2016 Table of Content Preface ...............................................................................................................................................................